T.C
SÜLEYMAN DEMİREL ÜNİVERSİTESİ
FEN BİLİMLERİ ENSTİTÜSÜ
YOĞUNLUK FONKSİYON TEORİ (DFT) METODU İLE KOLİN
BİLEŞİKLERİNİN TİTREŞİM ANALİZLERİ, OPTİMİZE
MOLEKÜL YAPILARI,
1H ve
13C NMR KİMYASAL
KAYMALARI
Mustafa KARAKAYA
Danışman: Prof. Dr. Fatih UCUN
DOKTORA TEZİ FİZİK ANABİLİM DALI
TEZ ONAYI
Mustafa KARAKAYA tarafından hazırlanan “Yoğunluk Fonksiyon Teori (DFT)
Metodu ile Kolin Bileşiklerinin Titreşim Analizleri, Optimize Moleküler Yapıları, 1
H ve 13C NMR Kimyasal Kaymaları” adlı tez çalışması aşağıdaki jüri
tarafından oy birliği / oy çokluğu ile Süleyman Demirel Üniversitesi Fizik Anabilim Dalı’nda DOKTORA TEZİ olarak kabul edilmiştir.
Danışman: Prof. Dr. Fatih UCUN
Süleyman Demirel Üniversitesi, Fizik Anabilim Dalı
Jüri Üyeleri:
Prof. Dr. Semiha BAHÇELİ
Süleyman Demirel Üniversitesi, Fizik Anabilim Dalı Doç. Dr. Hakan AKTAŞ
Süleyman Demirel Üniversitesi, Kimya Anabilim Dalı Doç. Dr. Ekrem ÇİÇEK
Mehmet Akif Ersoy Üniversitesi, Fizik Anabilim Dalı
Yrd. Doç. Dr. Ahmet TOKATLI
Süleyman Demirel Üniversitesi, Fizik Anabilim Dalı
Prof. Dr. Mehmet Cengiz KAYACAN Enstitü Müdürü
İÇİNDEKİLER Sayfa İÇİNDEKİLER………...………..i ÖZET.………...………...iv ABSTRACT...v TEŞEKKÜR……….………vi ŞEKİLLER DİZİNİ………vii ÇİZELGELER DİZİNİ………...……….………ix SİMGELER DİZİNİ………xi 1. GİRİŞ………1 2. KURAMSAL TEMELLER……….………...5 2.1. Molekül Yapı…………...5 2.2. Hamiltonien…...………...6
2.3. Born-Oppenheimer Yaklaşımı………...7
2.4. Hidrojen Molekülünün Değerlik-Bağı Yöntemiyle İncelenmesi...9
2.5. Çok Elektronlu Sistemler İçin Hesaplamalar………...17
2.5.1. Thomas-Fermi modeli………...18
2.5.2. Hartree-Fock metodu……….………..21
2.5.3. Hartree-SCF metodu………...25
2.5.4. Elektron korelasyon enerjisi………...29
2.5.5. Yoğunluk fonksiyon teorisi (DFT)…...30
2.5.6. B3LYP karma yoğunluk fonksiyonel teorisi………...32
2.6. Temel Setler………...33
2.7. Spektroskopi………...35
2.7.1. Enerjinin kuantizasyonu………38
2.7.2. Spektrum bölgeleri...40
2.7.2.1. Radyo frekans bölgesi...40
2.7.2.2. Mikrodalga bölgesi………40
2.7.2.3. İnfra-red (kızıl ötesi) bölgesi……….40
2.7.2.4. Görünür ve ultra-viyole bölgesi……….41
2.7.2.6. γ–ışını bölgesi………41
2.8. Çok Atomlu Moleküllerin Titreşimleri………41
2.8.1. Gerilme titreşimi………...42
2.8.2. Açı bükülme titreşimi………...42
2.8.3. Burulma titreşimi………..42
2.8.4. Düzlem dışı açı bükülme titreşimi………43
3. MATERYAL ve YÖNTEM………...44
3.1. Hesaplama Detayları………44
4. ARAŞTIRMA BULGULARI VE TARTIŞMA……….46
4.1. Kolin Halojenürler………...46
4.1.1. Temel hal konformasyonları……….46
4.1.2. Titreşim simetrileri………46
4.1.3. Optimize geometrik yapılar………..47
4.1.4. Titreşim frekansları………...49
4.1.5. NMR analizi………..52
4.2. Asetil Kolin Halojenürler……….55
4.2.1. Temel hal konformasyon analizi………...55
4.2.2. Optimize geometrik yapılar………..58
4.2.3. Titreşim frekansları………...61
4.2.4. Kimyasal kaymalar………...65
4.2.5. İnfrared ve Raman spektrum analizleri……….……..…………..70
4.3. Benzoil Kolin Klorür Molekülü………...73
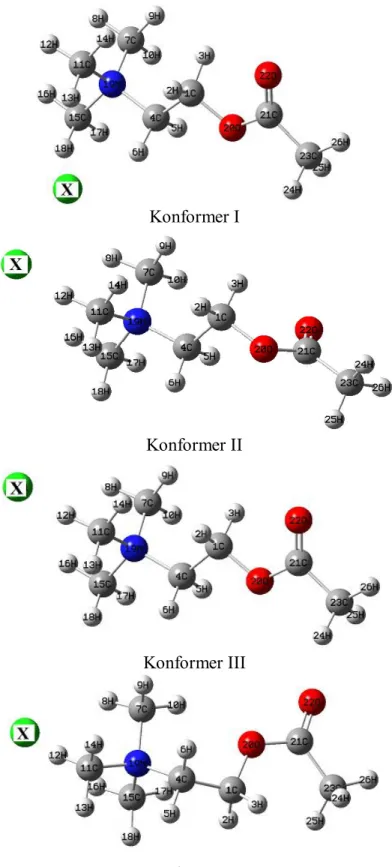
4.3.1. Temel hal konformasyon analizi………...………73
4.3.2. Optimize geometrik yapılar………..76
4.3.3. Titreşim frekansları………...79
4.3.4. NMR analizi………..………83
4.3.5. İnfrared ve Raman spektrum analizleri...………..85
4.3.6. Doğal bağ orbital (NBO) analizi………...87
4.3.7. Frontier molekül orbital (FMO) ve UV-görünür bölge spektroskopi analizleri….………...88
4.4.1. Temel hal konformasyon analizi………...………95
4.4.2. Optimize geometrik yapılar………..97
4.4.3. Titreşim frekansları………..…….99
4.4.4. NMR analizi………..………..110
4.4.5. İnfrared ve Raman spektrum analizleri……...………112
4.4.6. Doğal bağ orbital (NBO) analizi……….114
4.4.7. Frontier molekül orbital (FMO) ve UV-görünür bölge spektroskopi analizleri….……….116
4.4.8. Atomik yükler ve termodinamik parametreler………....120
4.4.9. Molekül elektrostatik potansiyel (MEP) analizi…...………..123
5. SONUÇ……….124
6. KAYNAKLAR...127
ÖZET Doktora Tezi
YOĞUNLUK FONKSİYON TEORİ (DFT) METODU İLE KOLİN BİLEŞİKLERİNİN TİTREŞİM ANALİZLERİ, OPTİMİZE MOLEKÜL YAPILARI, 1
H ve 13C NMR KİMYASAL KAYMALARI
Mustafa KARAKAYA Süleyman Demirel Üniversitesi
Fen Bilimleri Enstitüsü Fizik Anabilim Dalı
Danışman: Prof. Dr. Fatih UCUN
Bu tez çalışmasında, farklı temel setlerde yoğunluk fonksiyon teori (DFT) metodu ile bazı kolin bileşiklerinin mümkün konformerlerinin optimize molekül yapıları, titreşim frekansları, 1
H ve 13C nükleer manyetik rezonans (NMR) kimyasal kayma değerleri hesaplanmıştır. İlk kısımda, kolin halojenürler (F, Cl, Br) için kimyasal kaymalara ve titreşim frekanslarına, halojen katkılanmalarının, yani elektro negatifliğin etkileri araştırılmış ve yüksek frekans bölgelerinde azalan halojen elektronegatifliği ile frekansların arttığı görülmüştür. Düşük frekans bölgelerindeki örgü titreşimleri için ise durum tam tersi olmuştur. 1H kimyasal kaymaları,
δ(F)>δ(Cl)>δ(Br) sıralamasına sahipken, 13
C kimyasal kaymalarının
δ(Br)>δ(Cl)>δ(F) sıralamasına sahip olduğu görülmüş ve değişimler yorumlanmıştır.
İkinci kısımda asetil kolin halojenürlerde, rölatif enerjilerin ve ortalama titreşimsel sapmanın en düşük olduğu üç konformer yapı için teorik IR ve R spektrumları elde edilmiş ve ayrı ayrı olarak deneysel spektrumlarla iyi bir uyum sağlamadığı görülmüştür. Bundan dolayı bu üç konformere ait spektrumların aynı anda deneysel spektrumda bulunabileceği düşüncesiyle, spektrumların (IR ve R) toplamları çizilmiş ve bunların deneysel spektrumlarla çok daha iyi uyum sağladığı görülmüştür. Ayrıca üç konformerin ortalama NMR kimyasal kayma değerlerinin, ayrı ayrı konformerlere göre, deneysel verilerle uyumunun daha iyi olduğu görülmüştür. Son kısımda benzoil kolin klorür, kloro kolin klorür ve bromo kolin bromür için iki düşük enerjili konformerin teorik IR, R spektrumlarının üst üste çizimlerinin ve ortalama NMR kimyasal kayma değerlerinin deneysel verilerle karşılaştırılmasıyla çok daha iyi uyum sağladığı görülmüş ve iki konformerin deneysel spektrumda eş zamanlı bulunduğu sonucuna varılmıştır. Ayrıca, anyonların konumlarına göre belirlenen konformerler için toplam hiper konjuge enerjilerin birbirlerine çok yakın olması, konformerlerin optimize enerjilerinin de yakın olması gerektiğini desteklemiştir.
Anahtar Kelimeler: Yoğunluk fonksiyon teorisi (DFT), kolin bileşikleri, İnfrared
ABSTRACT Ph.D. Thesis
VIBRATIONAL ANALYSIS, OPTIMIZED MOLECULAR STRUCTURES,
1
H and 13C NMR CHEMICAL SHIFTS OF CHOLINE COMPOUNDS BY USING DENSITY FUNCTIONAL THEORY (DFT) METHOD
Mustafa KARAKAYA Süleyman Demirel University
Graduate School of Applied and Natural Sciences Physics Department
Supervisor: Prof. Dr. Fatih UCUN
In this thesis, the optimized molecular structures, vibrational frequencies, 1H and 13C nuclear magnetic resonance (NMR) chemical shifts of the possible conformers of some choline compounds have been calculated using DFT method at different basis set levels. In the first part, the electronegativity influence of the halogen substitutions on the vibrational frequencies and chemical shifts for choline halides (F, Cl, Br) have been investigated and it was seen that the calculated frequencies increase with decreasing electronegativity of the halide anion in the higher frequency regions. For the lattice vibrations in the lower frequency regions, the situation is vice versa. They were seen and commented that those of 13C chemical shifts are in the order
δ(Br)>δ(Cl)>δ(F), while 1
H chemical shifts have in the order δ(F)>δ(Cl)>δ(Br). In the second part, , the theoretical IR and R spectra for the three conformers of acetylcholine halides which have the low relative energies and mean calculated vibrational deviations were obtained. It was seen the experimental spectra do not fit well to the calculated spectra of the conformers, individually. So, the sum of the calculated spectra (IR or R) were drawn with the idea of that the spectra of these three conformers could simultaneously exist in one experimental spectrum and it was seen that these spectra better fit to the experimental spectra according to the individual conformers. It was also seen that the average values of chemical shifts of three conformers better fit to the experimental spectra according to the individual conformers. In the last part, it was seen that the average values of the calculated chemical shifts and the calculated IR and R spectra plotted together for two low-energy conformers of benzoylcholine chloride, chlorocholine chloride and bromocholine bromide fit well to experimental data and it was concluded that the experimental spectra are formed from the superposition of the spectra of these two conformers. Also, the total hyperconjugative energies are very near for the conformers determined relative to the location of the anions have supported that the two conformers of the compound should have close optimized energies.
Key Words: Density functional theory (DFT), choline compounds, Infrared spectra,
Raman spectra, NMR spectra, NBO analysis.
TEŞEKKÜR
Çalışmalarım sırasında her türlü desteğini aldığım, bilgi ve tecrübelerinden faydalandığım, ilgi ve yardımlarını esirgemeyen, değerli danışman hocam Sayın Prof. Dr. Fatih UCUN’a teşekkürlerimi sunarım. Ayrıca çalışmalarıma katkılarından dolayı bütün fizik bölümü hocalarıma ve maddi, manevi desteklerini esirgemeyen aileme teşekkür ederim. Saygılarımla…
Mustafa KARAKAYA ISPARTA, 2012
ŞEKİLLER DİZİNİ
Şekil 2.1. Hidrojen molekülünün Hamiltonienindeki uzaklıkların tanımı ……..…...8
Şekil 2.2. İki hidrojen çekirdeğinin 1s yörüngemsilerinin üst üste gelmesi………...11
Şekil 2.3. Eliptik koordinatlar………..…..……….12
Şekil 2.4. Hidrojen atomunun 1s yörüngemsilerinin üst üste gelme integrali S’nin R a değerine göre çizimi………..……….….12 0 Şekil 2.5. Coulomb integrali J ve değiş-tokuş integrali K’nın çekirdekler arasındaki uzaklığın fonksiyonu olarak çizimi.………..15
Şekil 2.6. Değerlik-bağı yöntemine göre H2 molekülünün çekirdekler arası potansiyel enerji eğrileri………..………...16
Şekil 2.7. Thomas-Fermi denkleminin çözümü………...…………...20
Şekil 2.8. y=Asinθ eğrisi……….36
Şekil 2.9. Bir P noktasının düzgün w açısal hızı ile dairesel hareketi terimlerinde bir sinüs eğrisinin açıklaması……….37
Şekil 2.10. dalga boylu ilerleyen bir dalga………..37
Şekil 2.11. E1 ve E2 enerji seviyeleri………..39
Şekil 2.12. Molekül titreşim türleri……...………..43
Şekil 4.1. Kolin halojenürlerin optimize molekül yapıları (X= F, Cl, Br)..…………46
Şekil 4.2. ChF’ye göre ChCl ve ChBr’nin hesaplanan titreşim frekansı sapmaları...52
Şekil 4.3. ChF’ye göre ChCl ve ChBr’nin hesaplanan kimyasal kayma sapmaları...53
Şekil 4.4. Asetil kolin halojenürlerinin tüm optimize konformerlerine ait molekül yapıları (X; F, Cl, Br)……..……….56
Şekil 4.5. AChF’nin sırasıyla A, B ve C olarak verilen konformer I, II ve III yapıları için hesaplanan IR ve R spektrumları...71
Şekil 4.6. AChCl’nin sırasıyla A, B ve C olarak verilen konformer I, II ve III yapıları için hesaplanan IR ve R spektrumları...72
Şekil 4.7. AChBr’nin sırasıyla A, B ve C olarak verilen konformer I, II ve III yapıları için hesaplanan IR ve R spektrumları...73
Şekil 4.8. BzChCl’nin konformerlerine ait molekül yapıları..………74
Şekil 4.9. Cl34–C11-N19-C4 torsiyon açısı üzerinde BzChCl’nin PES taraması…..75
Şekil 4.10. BzChCl-konformer I (A) ve konformer II (B) için hesaplanan Raman spektrumu…...……….….86
Şekil 4.11. BzChCl-konformer I (A) ve konformer II (B) için hesaplanan İnfrared spektrumu….………...………...…….87
Şekil 4.12. BzChCl-konformer I için frontier ve ikinci frontier molekül
orbitaller………... 89 Şekil 4.13. BzChCl’nin konformerleri için Mulliken tipi atomik dağılımları………91 Şekil 4.14. BzChCl-konformer I ve II için elektrostatik potansiyel değerlerine
göre elektron yoğunluk yüzeyi………..94 Şekil 4.15. ClChCl ve BrChBr’nin düşük enerjili iki konformeri için optimize
molekül yapılar (X=Cl,Br)…...……….96 Şekil 4.16. ClChCl’nin konformer I (A) ve konformer II (B) yapıları için
hesaplanan IR ve R spektrumları...………...113 Şekil 4.17. BrChBr’nin konformer I (A) ve konformer II (B) yapıları için
hesaplanan IR ve R spektrumları...………...114 Şekil 4.18. ClChCl’nin konformerleri için frontier ve ikinci frontier
molekül orbitaller……...………117 Şekil 4.19. BrChBr’nin konformerleri için frontier ve ikinci frontier
molekül orbitaller ...………118 Şekil 4.20. ClChCl ve BrChBr’nin düşük enerjili konformerleri için
Mulliken tipi atomik yük dağılımları………..…121 Şekil 4.21. ClChCl ve BrChBr’nin düşük enerjili konformerleri için
elektrostatik potansiyel değerlerine göre elektron yoğunluk yüzeyi…..123
ÇİZELGELER DİZİNİ
Çizelge 4.1 Cs nokta grubuna ait karakter çizelgesi………47
Çizelge 4.2. Kolin halojenürler için hesaplanan optimize geometrik yapı
parametreleri………...48 Çizelge 4.3. Kolin halojenürler için deneysel ve teorik titreşim frekansları………..50 Çizelge 4.4. Kolin halojenürlerin grup olarak teorik ve deneysel 1H ve
13C NMR kimyasal kaymaları……….53
Çizelge 4.5. Kolin halojenürlerin atomik olarak hesaplanan 1H ve 13C NMR
kimyasal kaymaları……….54 Çizelge 4.6. Asetil kolin halojenürlerin tüm konformerleri için 1[C(21)-O(20)
-C(1)-C(4)] ve 2[O(20)-C(1)-C(4)-N(19)] torsiyon açıları……….57
Çizelge 4.7. Asetil kolin halojenürlerin konformerleri için “elektronik ve sıfır nokta enerjileri” toplamı ve konformerler arasında
rölatif enerjiler, ortalama titreşimsel sapmalar………...57 Çizelge 4.8. Asetil kolin halojenürlerin deneysel ve en düşük enerjili
konformer I yapıları için optimize geometrik yapı parametreleri……...59 Çizelge 4.9. Asetil kolin halojenürlerin deneysel ve konformer I yapıları için
teorik titreşim frekansları………62 Çizelge 4.10. Asetil kolin halojenürlerin düşük enerjili konformerleri için
deneysel ve teorik kimyasal kayma değerleri………...67 Çizelge 4.11. BzChCl’nin konformer yapılarını tanımlayan torsiyon açıları……….75 Çizelge 4.12. BzChCl’nin konformerleri arasında rölatif enerji, ortalama
titreşimsel sapma ve elektronik enerji değerleri………...76 Çizelge 4.13. BzChCl’nin düşük enerjili konformerleri için hesaplanan
optimize geometrik yapı parametreleri……….77 Çizelge 4.14. BzChCl’nin düşük enerjili konformerleri için deneysel ve teorik
titreşim frekansları………80 Çizelge 4.15. BzChCl’nin düşük enerjili konformerleri için deneysel ve
tetrametilsilan (TMS)’a göre hesaplanan 1H ve 13C izotropik
kimyasal kayma değerleri……….84 Çizelge 4.16. BzChCl’nin düşük enerjili iki konformeri için aşırı konjuge
etkileşimler (kcalmol-1)……….88
Çizelge 4.17. BzChCl’nin konformerleri için hesaplanan HOMO, LUMO,
Çizelge 4.18. BzChCl’nin düşük enerjili iki konformeri için TD-DFT metodu ile hesaplanan dalga boyları λ(nm), uyarılma enerjileri (eV)
ve osilatör siddet (f) değerleri………...90 Çizelge 4.19. BzChCl’nin düşük enerjili iki konformeri için atomik yükler………..92 Çizelge 4.20. BzChCl’nin düşük enerjili iki konfermeri için termodinamik
özellikler………...93 Çizelge 4.21. ClChCl ve BrChBr’nin konformerleri için hesaplanan
torsiyon açıları……….96
Çizelge 4.22. ClChCl ve BrChBr’nin konformerleri için elektronik enerjiler
ve konformerler arasında rölatif enerji değerleri………..97 Çizelge 4.23. ClChCl ve BrChBr’nin düşük enerjili konformerleri için teorik,
optimize geometrik yapı parametreleri……….98 Çizelge 4.24. ClChCl ve BrChBr’nin konformer I yapıları için deneysel ve
teorik titreşim frekansları………101
Çizelge 4.25. ClChCl ve BrChBr’nin konformer II yapıları için deneysel ve
teorik titreşim frekansları………106
Çizelge 4.26. ClChCl ve BrChBr’nin konformerleri için deneysel ve teorik
13C ve 1H NMR izotropik kimyasal kaymaları...111 Çizelge 4.27. ClChCl ve BrChBr’nin konformerleri için aşırı konjuge
etkileşimler………..116
Çizelge 4.28. ClChCl ve BrChBr’nin konformerleri için hesaplanan HOMO,
LUMO, HOMO-1, LUMO+1 enerji değerleri………119 Çizelge 4.29. ClChCl ve BrChBr’nin konformerleri için TD-DFT/B3LYP
metodu ile hesaplanan dalga boyları λ(nm), uyarılma enerjileri(eV) ve osilatör şiddet (f) değerleri……….120 Çizelge 4.30. ClChCl ve BrChBr’nin konformerlerine ait optimize geometrilere
göre Mulliken tipi atomik yükler………121 Çizelge 4.31. ClChCl ve BrChBr’nin konformerleri için termodinamik
SİMGELER DİZİNİ
H Hamiltonien operatör
Z Atom numarası
jk
r
∆ İki parçacık arasındaki uzaklık
2
a
∇ , a çekirdeklerinin bulundukları yerlere göre Laplacian işlemcisi
2
b
∇ b çekirdeklerinin bulundukları yerlere göre Laplacian işlemcisi
1s a Elektronu a çekirdeği üzerine yoğunlaşmış 1s hidrojen yörüngemsisi 1s b Elektronu b çekirdeği üzerine yoğunlaşmış 1s hidrojen yörüngemsisi
J Coulomb integrali
K Değiş-tokuş integrali
1
2E Ys alıtılmış iki hidrojen atomunun taban durum enerjisi
TF Thomas-Fermi teorisi
( )
r ρ Elektron yoğunluğu( )
r φ Elektrostatik potansiyel( )
V r Potansiyel enerjiHF Hartree-Fock yaklaşımı
F Fock operatörü
RHF Sınırlandırılmış Hartree-Fock yaklaşımı UHF Sınırlandırılmamış Hartree-Fock yaklaşımı SCF Öz Uyumlu Alan Metodu (Self-Consistent Field)
etkin
V Etkin potansiyel enerji etkin
H Etkin Hamiltonien operatörü
exact
E Tam enerji exact
ψ Rölativistik olmayan Hamiltonienin tamdalga fonksiyonu corr
E Korelasyon enerjisi
DFT Yoğunluk Fonksiyon Teorisi XC
XC
E Değiş-tokuş korelasyon enerjisi C E Korelasyon enerjisi X E Değiş-tokuş enerjisi S E Slater fonksiyonel VWN
E Vosko, Wilk ve Nusair (1980) tarafından tanımlanan fonksiyonel B
E Becke (1988) tarafından tanımlanan fonksiyonel LYP
E Lee, Yang ve Parr (1988) tarafından tanımlanmış fonksiyonel
i
ψ Moleküler orbital
µ
φ Atomik orbital i
cµ Moleküler orbital açılım katsayıları ChF Kolin florür
ChCl Kolin klorür ChBr Kolin bromür AChF Asetil kolin florür AChCl Asetil kolin klorür AChBr Asetil kolin bromür BzChCl Benzoil kolin klorür ClChCl Kloro kolin klorür BrChBr Bromo kolin klorür TMS Tetrametilsilan
R2 Lineer korelasyon katsayısı RMSE Ortalama hata karenin karekökü MAE Ortalama mutlak hata
NBO Doğal bağ orbitali
LP Bağ yapmamış elektron çifti FMO Frontier molekül orbital
MEP Molekül elektrostatik potansiyel PES Potansiyel enerji yüzeyi
1. GİRİŞ
Çok elektronlu sistemlerde sayısal çözümlemeler için çeşitli yöntemler kullanılmaktadır. Bunlar; ab initio yöntemleri, yarı-ampirik yöntemler, yoğunluk fonksiyonel yöntemleridir. Bu yöntemlerle Schrödinger denklemi, tutarlı sonuç elde edinceye kadar tekrarlanarak çok sayıda çözümlenir. Bu hesaplamalar çok karmaşık ve çözümü zaman aldığı için ticari paket yazılımlar tarafından yapılmaktadır. Bu yazılımlar fortran programlama dili kullanılarak hazırlanır ve bu yazılımlardan bazıları Gaussian, Gamess, Molpro, Cache’ dir.
Ab initio, latince “başlangıçtan itibaren, sıfırdan itibaren” olarak tanımlanır. Ab initio yöntemi de “sıfırdan teorik yöntemler” olarak adlandırılır. Bunun sebebi, ab initio yöntemleri hiçbir yaklaşımı esas almadan tamamen Hamiltonieni kullanmasıdır. Ayrıca bu yöntemde hiçbir denel veri de kullanılmaz. Yarı-ampirik yöntemlerde, ihtimallerin uygulanmasıyla integrallerde kolaylıklar sağlanır. Yarı-ampirik yöntemlerde hesaplama süresi, ab initio hesaplamalarına göre daha kısadır. Fakat sonuçlar ab initio yöntemlerde daha hassastır ve 100 atomlu sistemlere kadar kullanılabilir. İncelenen yapılar büyüdükçe hesaplamalar da zorlaşır ve bilgisayar yazılımları işlevsiz duruma düşebilir (Pilar, 1990). Schrödinger denklemi iki elektronlu atomlar için tam olarak çözümlenemediği için belli başlı yaklaşım metotları kullanılmaktadır. Uygun varyasyonel hesapların yerine getirilmesiyle iki elektronlu atomların enerji seviyeleri ve dalga fonksiyonları için doğru sonuçlar elde edilebilir. Fakat atomik sistemde elektron sayıları arttığında bu metotların kullanımı giderek zor hale gelmektedir. Bu yüzden çok elektronlu atomik ve moleküler sistemler üzerinde çalışmak için bazı farklı genel metotlar geliştirilmiştir (Erkoç and Uzer, 1996).
Elektronun çekirdekten uzaklığı r ise her bir elektronun, küresel simetrik bir potansiyel V(r)’ de, bağımsız hareketi düşünülerek pertürbe olmamış bir sistem elde edilebilir. Burada V(r) potansiyeli, çekirdeğin oluşturduğu çekme potansiyeli ile elektronla diğer elektronlar arasındaki ortalama itme potansiyelinin toplamıdır. Genel çözüm için önce kaba bir itme potansiyeli kullanılır ve dalga fonksiyonu elde edilir.
Daha sonra dalga fonksiyonu kullanılarak bu itme potansiyeli düzeltilir. Elde edilen bu düzeltilmiş potansiyele öz uyumlu (self-consistent) potansiyel denir. Başlangıçtaki potansiyel ile bitiş potansiyeli arasında fark kalmayıncaya kadar bütün bu işlemlere devam edilir. Hartree teorisi ve Thomas-Fermi (TF) teorisi, öz uyumlu yaklaşımı esas alan önemli teorilerdir (Şahin ve Kurucu, 2005). Bölüm 2’de bu teorilerle birlikte ab initio Hartree-Fock (HF) ve yoğunluk fonksiyonel teori (DFT) metotlarına da yer verilmiştir.
Bu çalışmada hesaplamalar kolin bileşikleri üzerine olduğu için, bu konuda mevcut çalışmalar hakkında bilgi vermek istedik. Kolin bileşikleri [(CH3)3NCH2COOH]+,
sıra dışı radyasyon duyarlılıkları ve biyolojik sistemlerde sık görülmesi sebebiyle dikkat çekmektedir. Bu yapılar kompleks lipidlerdir ve trans metil etken maddesi olarak görev alırlar (Fischer et al., 1970). Köksal ve Bahçeli (1983), NMR spektroskopisi ile bazı kolin ve asetil kolin halojenürlerde spin-örgü durulmasına spin difüzyonunun ve metil grup dönmelerinin etkilerini incelemiştir. Ayrıca Akın ve Harmon (1994), yine NMR spektroskopisiyle sulu çözeltilerde kolin ve asetil kolin halojenürlerin hidrasyonu üzerindeki anestetiklerin etkileri araştırmışlardır. IR spektroskopisi ile kolin bromür ve kolin iyodürün yüksek sıcaklık fazları ile ilgili çalışmalar mevcuttur (Harmon et al., 1986). Yine bazı araştırmacılar tarafından kolin ve asetil kolin halojenürlerin daha düşük hidratları üzerine NMR ve IR spektroskopik çalışmalar yapılmıştır (Harmon and Avci, 1984; Harmon et al., 1985; 1991). X ışını kırınımı metodu kullanılarak kolin klorürün kristal yapısı üzerine incelemeler de literatürde mevcuttur (Senko and Templeton, 1960; Hjortas and Sorum, 1971).
Asetil kolin [CH3COOCH2CH2N+(CH3)3] bileşikleri, canlılar için sinirsel iletimde
merkezi bir role sahiptir. Kolin ve asetil kolin türevleri, donanım performası açısından programlardaki hesaplama zorlukları sebebiyle, moleküler karmaşıklığa sahip moleküller olarak bilinir. Bu sebeple çok farklı temel setlerin kullanımıyla kolin ve asetilkolinlerin titreşim frekanslarını analiz etmek bir çok araştırmacının dikkatini çekmiştir (Derreumaux et al., 1989; Davies et al., 2003; Karakaya et al., 2010). Asetilkolinin potansiyel enerji yüzeylerinde minimum enerjili konformerlerini
2004). Bu çalışmalar göstermiştir ki; düşük enerjili konformerler, molekülün geri kalan kısmına göre asetoksi grubunun oryantasyonuna uygun görünmektedir. Potansiyel enerji yüzeylerinde farklı minimal enerjili konformer yapılar, asetilkolinin muskarinik ve nikotinik aktiviteleri ile ilgilidir (Munoz-Caro et al., 2005).
Marino et al. (2001), sulu çözelti ve vakumda asetilkolinin moleküler yapısı ve konformasyonel davranışı ile ilgili çalışmalar yapmışlardır. Bu araştırmacılar moleküler mekanik tabanlı hesaplamalar yardımıyla beş düşük enerjili konformer yapısı bulmuşlardır. Başka bir çalışmada, asetil kolin molekülünün ab initio verileri, en kararlı konformerin iki önemli torsiyon açısıyla (1; C-C-O-C ve 2; N-C-C-O)
trans-gauche dizilişi olduğunu göstermiştir (Caillet et al., 1978; Segall, 1998). Asetil kolin klorür kristalinde trans-gauche (1= -166.9ve 2 = 84.7o), asetilkolin bromür
kristalinde gauche-gauche (1= 78.9 ve 2= 78.4o) ve asetilkolin iyodür kristalinde
gauche-gauche (1= ±83 ve 2= ±89o) yapısı vardır (Herdklotz and Sass, 1970;
Svinning and Sorum, 1975; Jagner and Jensen, 1977; Frydenvang and Jensen, 1996). Asetilkolin bromür için teorik çalışmalar göstermiştir ki; deneysel gauche-gauche konformasyonu farklı bir gauche-gauche yapıya uyan gerçek minimum konformasyonunun yaklaşık 2 kcal/mol üzerindedir (Pullman and Courriere, 1972). Yukarıdaki tartışmalardan da anlaşıldığı gibi asetilkolinin temel hal konformasyonu, yapılan deneysel durumlara bağlı değişmektedir.
Bu çalışmada, farklı temel setlerle yoğunluk fonksiyon teori (DFT/B3LYP) metodu ile belirtilen kolin bileşiklerinin konformerleri için optimize moleküler yapı parametreleri, titreşim frekansları, 1H ve 13C nükleer manyetik rezonans (NMR) kimyasal kayma değerleri hesaplanmıştır. Kolin bileşiklerinin İnfrared, Raman ve NMR spektrumlarının deneysel ve teorik değerlerinin karşılaştırmaları, birden fazla optimize minimum enerjili konformerlere ait spektrumların eş zamanlı olarak deneysel spektrumlarında bulunabileceğini göstermiştir. Yani, bileşiklerin temel halde birden fazla konformerinin aynı anda bulunabileceği sonucuna varılmıştır. Doğal bağ orbital (NBO) analizi de birden fazla konformerin temel halde eş zamanlı bulunabileceği fikrini desteklemiştir. Teorik frekansların titreşim kipi atamaları, potansiyel enerji dağılımı (PED) yaklaşımı ile yapılmıştır. Tüm konformerler için
teorik optimize geometrik yapı parametrelerinin (bağ uzunluğu, bağ açıları), titreşim frekanslarının, 1H ve 13C NMR kimyasal kayma değerlerinin ilgili deneysel verilerle iyi bir uyum içinde olduğu görülmüştür.
Ayrıca çalışmamızdaki bazı kolin bileşiklerinin tüm konformerlerinin ilave olarak termodinamik özellikleri, frontier molekül orbital (FMO) analizleri, molekül elektrostatik potansiyel (MEP) analizleri yapılmıştır. Ayrıca, zamana bağlı yoğunluk fonksiyon teori (TD-DFT) metoduyla, UV görünür bölge spektrum analizi olarak konformerlerin dalga boyları, uyarılma enerjileri ve osilatör şiddetleri hesaplanmıştır.
2. KURAMSAL TEMELLER
2.1. Molekül Yapı
Kuantum mekaniği, hem parçacık özelliği hem de dalga özelliği gösteren elektronları incelemektedir. Schrödinger denklemi,
2 2 2 , , 8 2 r t h ih V r t m t (2.1)şeklinde tanımlanır. Bu denklemde dalga fonksiyonu, m parçacığın kütlesi, h Planck sabiti ve V, parçacığın hareket ettiği potansiyeldir. Dalga fonksiyonu ile kompleks eşleniğinin çarpımı, parçacığın olasılık dağılımı olarak açıklanır.
Parçacıkların enerjisi ve diğer birçok özellikleri, uygun sınır şartlarına bağlı olarak Schrödinger denkleminin çözümü ile elde edilebilir. Potansiyel V, zamana bağlı bir fonksiyon değilse değişkenlerin ayrılması olarak bilinen bir matematiksel yöntemin kullanılmasıyla Schrödinger denklemi basitleştirilebilir. Dalga fonksiyonu, zamanın ve uzayın fonksiyonu olarak
r t,
r
t (2.2)
şeklinde yazılır. Problemler için zamandan bağımsız Schrödinger denklemi ise
H r E r (2.3)
olarak verilir. Burada, E parçacığın enerjisi, H ise Hamiltonien operatördür ve
2 2 2 8 h H V m (2.4)
eşitliği ile verilir. Denklem (2.3) için çeşitli çözümler, molekülün farklı durumlarına uymaktadır. Bunlardan en düşük enerjili olanı, temel hal olarak adlandırılır. Denklem (2.3), parçacık hızlarının ışık hızına yaklaştığı durumlarda geçerlidir.
2.2. Hamiltonien
Moleküler bir sistem için , molekül içindeki elektronlar ve çekirdeğin konumlarını içeren bir dalga fonksiyonudur. Her bir parçacık için elektron ve çekirdeğin konumlarını tanımlayan bileşke vektörler sırasıyla burada r Ri, I
olarak verilmiştir. Hamiltonien, kinetik ve potansiyel enerji terimlerinden oluşmaktadır ve
H TV (2.5)
olarak ifade edilir. Kinetik enerji, molekülde tüm parçacıkların kinetik enerjilerinin toplamıdır ve 2 2 2 2 2 2 2 2 1 8 k k k k k h T m x y z
(2.6)eşitliği ile verilir. Potansiyel enerji ise, yüklü yapıların her bir çifti arasındaki Coulomb itmesidir ve 0 1 4 j k j k j jk e e V r
(2.7)ile ifade edilir. Burada rjkiki parçacık arasındaki uzaklıktır. ej, e ise j ve k k
parçacıklarının yükleridir. Z atom numarası olmak üzere –e, bir elektronun yükü ve
+Ze çekirdek yüküdür. Böylece,
2
2 2
1 elektronlarçekirdekler elektronlar çekirdekler
I J
I Z Z e
Z e e
olarak verilir. Burada birinci terim çekirdek çekimine, ikinci terim elektron-elektron itmesine ve üçüncü terim ise çekirdek-çekirdek itmesine aittir.
Kuantum kimyasının temel denklemleri, belirli sabitlerin yok edilmesiyle yani sadeleştirmeyle tasarlanan birimlerle ifade edilir. Uzunluğun atomik birimi, Bohr
yarıçapıdır ve 2 0 0 2 2 0.52917725 4 e h a A m e (2.9)
eşitliği ile yazılır. Koordinatlar, a bohr yarıçapına dönüştürülebilir, enerjiler 0
hartree şeklinde ölçülür. Hartree, 1 bohr olarak aralarında uzaklık olan iki elektron
arasındaki Coulomb itmesidir ve
2 0
1 hartree e a
(2.10)
şeklinde yazılır. Kütleler de elektron kütle birimi ifadesiyle yani m olarak verilir e 1 (Foresman and Frisch, 1996).
2.3. Born-Oppenheimer Yaklaşımı
Hidrojen molekülü için Hamiltonien işlemcisi
2 2 2 2 2 2 2 2 1 2 0 1 0 1 2 2 2 2 2 0 2 0 2 0 12 0 ˆ 2 2 4 4 4 4 4 4 4 a b a b a b Ze Ze H M m r r Ze Ze e e r r r R (2.11)olarak verilir. Burada M hidrojen çekirdeğinin kütlesi, m elektronun kütlesi, Z çekirdeğin atom numarasıdır. 2
a
, 2
b
ise a ve b çekirdeklerinin bulundukları yerlere göre Laplacian işlemcileridir (Yani 2
1
ve 2 2
, 1 ve 2 elektronlarının yerlerine göre Laplacian işlemcileridir).
Şekil 2.1. Hidrojen molekülünün Hamiltonienindeki uzaklıkların tanımı (Köksal ve Köseoğlu, 2006)
Burada çekirdeklerin kütleleri elektron kütlelerinden çok daha ağır olduğundan elektronlara göre çekirdekler hareketsiz kabul edilmektedir. Bu nedenle Denklem (2.11)’de 2a ,2b ihmal edilir ve
2 2 2 2 2 2 1 2 0 1 0 1 0 2 2 2 2 2 0 2 0 12 0 ˆ 2 4 4 4 4 4 4 a b a b Ze Ze Ze H m r r r Ze e Z e r r R (2.12)şeklinde yazılabilir. Çekirdeğin hareketinin ihmal edildiği bu yaklaşıma Born-Oppenheimer yaklaşımı denir. Molekülün başka biçimlenmesi hallerinde, uzaklıklar değiştirilerek her durumda Denklem (2.12) çözülür ve molekül için enerji düzeyleri çizilir. Buna göre molekülün denge durumuna karşılık gelen biçimlenmesi, potansiyel eğrisi veya yüzeyinin minimum noktasına karşılık gelir.
2.4. Hidrojen Molekülünün Değerlik-Bağı Yöntemiyle İncelenmesi
Kimyasal bağın ve özellikle H2 molekülünün kararlı oluşun ilk tatmin edici yaklaşımı
Heitler ve London (1927) tarafından verilmiştir. Daha sonra bu yöntem karmaşık moleküllere genişletilmiş ve bugün değerlik-bağı yöntemi olarak bilinmektedir. Burada helyum atomundaki gibi spin dikkate alınmayıp daha sonra probleme eklenecektir.
İki elektronlu sistemin dalga fonksiyonu, uzaysal ve spin kısımlarının çarpımı şeklinde yazılmaktadır. Hidrojen atomları kendi taban durumunda ve birbirinden yeterince uzakta iseler iki hidrojen atomunun dalga fonksiyonu için
1 1sa 1 1sb 2
(2.13) şeklinde yazılır. Burada 1s , elektronu a çekirdeği üzerine yoğunlaşmış 1s hidrojen a
yörüngemsisini ve 1s de elektronu b çekirdeği üzerine yoğunlaşmış hidrojen b
yörüngemsisini göstermektedir. a çekirdekleri, 1s yörüngemsilerinin tanımlandığı küresel koordinatların orijinleridir. Elektronlar birbirlerinden ayırt edilemez oldukları için yukarıdaki denkleme benzer şekilde
2 1sa 2 1sb 1
(2.14) bağıntısı yazılır. Deneme dalga fonksiyonu ve 1 nin bileşimi şeklinde ele 2
alınırsa
1 1 2 2
c c
(2.15) olur. Buna göre determinant biçimindeki kalıcı determinant,
1 1 1 11 11 12 12 2 12 12 22 22 H ES H ES H ES H ES (2.16)
olarak yazılır. Burada 1s yörüngemsileri normalize olduğundan
11 1 2 1 2 1 1 1 2 1 1 1 2 1 1 1 1 1 2 1 2 1 a b a b a a b b S s s s s d d s s d s s d
(2.17)sonucunu verir. Aynı şekilde S dir. 11 1
12 1 2 1 2 1 1 1 2 1 2 1 1 1 1 1 1 1 2 1 2 1 a b a b a b b a S s s s s d d s s d s s d
(2.18)şeklinde yazılır. Elektronlar ayırt edilemez parçacıklar olduğu için burada her iki integral de aynıdır. 1 elektronunun1s ve 1a s de bulunması birbirinden ayıt edilemez. b Sonuçta
2 12
S S ve S
1sa
1 1sb
1 d1 (2.19) dir. Bu integral Denklem (2.17)’deki integralden farklıdır. Çünkü bu integralde 1s yörüngemsilerinden birisi a ve diğeri b çekirdeği üzerine merkezlenmiştir. Şekil 2.2’de bu integraller verilmiştir. Denklem (2.19)’daki integralin sıfırdan farklı olması için 1s ve 1a s nin de sıfırdan farklı olması gerekir. S integralinin büyüklüğü 1b s ve a1s fonksiyonlarının üst üste gelme miktarına bağlıdır. Bu nedenle bu integraller üst b
üste gelme integrali olarak adlandırılır. Bu integral Şekil 2.2’de görüldüğü gibi çekirdekler arasındaki uzaklık R ye bağlıdır ve Şekil 2.3’te belirtilen eliptik koordinatlarda hesaplanır. Bu koordinatlarda değişkenler
rarb
R,dir. Burada rarb, R den küçük olamayacağı için ra rb de R yi aşamadığı için 1 , 1 1, 02 olarak yazılır.
Şekil 2.2. İki hidrojen çekirdeğinin 1s yörüngemsilerinin üst üste gelmesi
0
0 1 2 1 2 3 3 3 3 0 0 1 Zr aa , 1 Zr ab a b s Z a e s Z a e eşitlikleriyle birlikte
0 2 1 3 3 3 2 2 0 0 1 1 1 1 1 8 Z R a a b S s s d Z a d d d R e
(2.20)olarak yazılır. Hidrojen atomu için Z=1 dir. Denklem (2.20)’daki diğer integraller hesaplandığında,
2 0 0 0 1 1 3 ZR a S ZR a ZR a e (2.21)elde edilir. Şekil 2.4’te Denklem (2.20), Z=1 için R a değerinin fonksiyonu olarak 0
Şekil 2.3. Eliptik koordinatlar
Şekil 2.4. Hidrojen atomunun 1s yörüngemsilerinin üst üste gelme integrali S’nin
0
R a değerine göre çizimi
Denklem (2.16)’da verilen determinantın diğer elemanları hesaplandığında,
11 1 2
ˆ 1 1 1 2 ˆ1 1 1 2
a b a b
H
s s H s s d d (2.22) olarak yazılır. H , Denklem (2.12)’de Z=1 yerine yazıldığında elde edilen ˆHamiltoniendir. Denklem (2.22)’de 1s yörüngemsileri hidrojen atomunun özfonksiyonlarıdır ve
2 2 4 2 1 2 2 0 1 0 2 2 4 2 1 1 1 1 2 4 a a 8 a e me s s m r h e me (2.23)eşitlikleri yazılır. Öte yandan 4 2 2 2 0 0 1 2 12 1 1 1 1 ˆ 1 (1)1 (2) 1 (1)1 (2) 4 4 a b a b b a me e H s s s s h r r r R (2.24) olduğu için 11 2 1s H E J (2.25) olarak yazılır. Burada E bir hidrojen atomunun enerjisidir. J integrali 1s
2 2 2 1 2 0 1 2 12 1 1 1 1 1 (1) 1 (2) 4 a b a b e J s s d d r r r R
(2.26)olarak yazılır ve 1s yörüngemsilerinin normalize olduğu şekliyle
2 2 2 1 2 0 1 2 2 2 2 2 1 2 0 12 0 1 (1) 1 (2) 4 1 (1) 1 (2) 4 4 a b b a a b s s e J d d r r s s e e d d r R
(2.27)yazılır. Bu denklemde ilk terim 1. elektronun a çekirdeği etrafındaki yük yoğunluğunun b çekirdeği ile Coulomb etkileşme potansiyelini, ikinci terim 2. elektronun b çekirdeği etrafındaki yük yoğunluğunun a çekirdeği ile Coulomb etkileşme potansiyelini temsil etmektedir. Üçüncü terim iki elektronun yük dağılımlarının karşılıklı Coulomb etkileşmesi, dördüncü terim ise çekirdeklerin kendi aralarındaki Coulomb etkileşme terimidir. Denklem (2.27)’de J, Coulomb integrali olarak adlandırılır ve çekirdekler arasındaki uzaklığa bağlıdır. Denklem (2.22)’de 1 ve 2 elektronlarının yerleri değiştirildiğinde H değişmediği için 11 H11 H22 dir. H 12
12 1 2 ˆ 1 1 1 2 ˆ1 1 1 2 a b b a H
s s H s s d d (2.28) ifadesi yazılır ve
2 2 4 2 1 2 2 0 1 0 2 2 4 2 2 2 2 0 2 0 1 1 1 1 2 4 8 1 2 1 2 2 4 8 b b b a a a e me s s m r h e me s s m r h (2.29) eşitlikleriyle 4 2 2 2 2 2 2 0 0 1 0 1 0 12 0 ˆ 1 (1)1 (2) 1 (1)1 (2) 4 4 4 4 4 b a b a a b me e e e e H s s s s h r r r R (2.30)yazılır. Bu denklem soldan 1 (1)1 (2)sa sb ile çarpılır ve her iki elektronun koordinatları cinsinden integrali alınırsa
2 12 2 1s
H E S K (2.31)
elde edilir. Sonuçta
2 1 2 0 1 2 12 1 1 1 1 1 (1)1 (1) 1 (2)1 (2) 4 a b a b a b e K s s s s d d r r r R
(2.32)bağıntısı yazılabilir. Burada J’deki
1 (1)sa
2 yerine
1 (1)1 (1)sa sb
çarpanı göze çarpmaktadır. Bu durum Denklem (2.15)’te sunulan tipte bir dalga fonksiyonu kullanılmasından kaynaklanılmaktadır. Dalga fonksiyonu, elektronların birbirinden ayırt edilemediği gerçeğini kullanmaktadır. Burada K, elektronların iki çekirdek arasındaki değiş-tokuşundan kaynaklandığı için değiş-tokuş integrali olarakŞekil 2.5. Coulomb integrali J ve değiş-tokuş integrali K’nın çekirdekler arasındaki uzaklığın fonksiyonu olarak çizimi. J ve K’nın değişkenleri atomik birimler
cinsinden ifade edilmiştir
Gerekli hesaplamaların ardından Denklem (2.16) ile verilen determinant,
2 2 1 1 2 2 1 1 2 2 2 2 s s s s E J E E S K ES E S K ES E J E (2.33)
şeklinde yazılır. Bu determinantın açılımıyla
1 2 2 1 s J K E E S (2.34)
eşitliği yazılır. Burada 2E , yalıtılmış iki hidrojen atomunun taban durum enerjisi 1s
olduğundan birbirinden ayrı iki hidrojen atomuna göre H2 molekülünün enerjisi
2 1 J K E S (2.35)
olarak yazılır. Bu denklemin çekirdekler arasındaki uzaklığın fonksiyonu olarak çizimi Şekil 2.6’da verilmiştir.
Şekil 2.6. Değerlik-bağı yöntemine göre H2 molekülünün çekirdekler arası potansiyel
enerji eğrileri
E ve E enerjilerine karşılık gelen dalga fonksiyonları Denklem (2.16)’daki determinant ifadesinden,
1 11 11 2 12 12 1 12 12 2 22 22 0 0 c H ES c H ES c H ES c H ES (2.36)şeklinde verilen denklemlerde E ve E yerleştirilerek bulunur. Bu denklemde birinci ifade
2 2
1 2 1s 2 2 1s 0
c E JE c E S KE S (2.37)
olarak yazılır. E’nın Denklem (2.34)’deki değeri yerine yerleştirilmesiyle c1c2
olur. Böylece dalga fonksiyonu
1 1 2 c
(2.38)
şeklinde yazılır. Burada ve 1 , Denklem (2.13) ve (2.14)’te verilmiştir. Burada 2
1
2 2 1 1 2 1 1 c S ve
1 1 2 2 1 2 1 c S (2.39)bulunur. Sonuçta H2 molekülünün E değerine karşılık gelen dalga fonksiyonu,
2
1 2
1 2
1 2 1 S (2.40)ve aynı işlemler Edeğeri için yapılırsa
2
1 2
1 2
1 2 1 S (2.41)sonucu elde edilir. Yukarıdaki denklemlerde + ve – işaretleri, elektronların uzaysal koordinatları değiştirildiğinde fonksiyonların sırasıyla simetrik ve antisimetrik olduğunu ifade etmek için kullanılmıştır. Şekil 2.5’te K’nın negatif olduğu görülmektedir. Negatif değerler kararlılığı temsil eder ve değiş-tokuş integrali hidrojen molekülünün kararlılığını sağlar. Değiş-tokuş integrali tamamen kuantum mekaniksel bir nicelik olduğu için kimyasal bağın kararlılığı da kuantum mekaniksel bir olaydır. Şekil 2.6’da, Denklem (2.35)’de verilen E ve E’nin çizimleri verilmiştir. Burada E, kararlı bir kimyasal bağ göstermektedir. Çünkü enerji, yalıtılmış iki hidrojen atomunun enerjisinden daha küçüktür. Eeğrisi, atomlar arası etkileşmeyle oluşan ve kimyasal bir bağ oluşturduğunu ortaya koyan tipte bir eğridir (Köksal ve Köseoğlu, 2006).
2.5. Çok Elektronlu Sistemler İçin Hesaplamalar
Giriş bölümünde bahsettiğimiz gibi Schrödinger denklemi çok elektronlu atomlar için tam olarak çözümlenemez. Bunun için belli başlı yaklaşım metotları
kullanılmalıdır. Bu bölümde Thomas-Fermi (TF) teorisi, Hartree-Fock (HF) metodu, yoğunluk fonksiyon teorisi (DFT) ve B3LYP karma yoğunluk fonksiyonel teori gibi öz uyumlu yaklaşımı esas alan önemli teorilere yer verilmiştir.
2.5.1. Thomas-Fermi modeli
Thomas-Fermi (TF) modeli çok elektronlu atomların temel hal için istatistiksel ve yarı klasik düşünceye dayanır. Bu modelde, sistemin N elektronları temel durumdaki bir Fermi elektron gazı gibi ele alınır. Fermi elektron gazı, sonsuzda sıfır olan merkezi bir potansiyel V(r) tarafından uzayın bir bölgesinde sınırlandırılmıştır.
TF modelinde amaç, potansiyel V(r) ve elektron yoğunluğu
r ’nin hesaplanması için bir metot sağlamaktır. Burada sistemin toplam enerjisi,
FEE V r (2.42)
şeklinde yazılır. Ayrıca Fermi elektron gazı modelinde,
2 2 3 2 3 2 F E m (2.43)olarak verilir. Denklem (2.42) ve (2.43) birleştirildiğinde,
3 2 3 2 2 2 1 2 3 m r E V r (2.44)sonucu elde edilir. Burada V E için
r 0 dır. Elektrostatik potansiyel
r ve potansiyel enerji V r
arasındaki bağıntı,olarak tanımlanabilir. 0 1E e
eşitliği ile
r
r 0 (2.46)
bağıntısı yazılabilir. Sonuç olarak,
3 2 3 2 2 2 1 2 , 0 3 0, 0 m r e r için r için (2.47)bağıntıları elde edilir. Verilen bir yük yoğunluğu için Poisson’s denklemi,
2
r e r
(2.48)
şeklindedir. Denklem (2.48)’de Denklem (2.47) yerine konulursa,
3 2 3 2 2 2 2 2 2 , 0 3 0, 0 e m r e r için r için (2.49)elde edilir. Ayrıca 1 r
r Ze şeklinde tanımlanan fonksiyon ve xr b
eşitliklerinde,
2 3 7 3 3 1 3 0 2 b a Z (2.50)3 2 3 , 0 0, 0 Z için b x için (2.51)
şeklinde elde edilir. 2
1 r d
2r
r dr2 bilinen ifadeyle,2 3 2 2 1 d dx x (2.52)
eşitliği çıkarılır. Bu denklem Thomas-Fermi denklemi olarak bilinir. Burada 0 için d2 dx2 ve 0
0 1’dir. Nötr atom çözümü için
0’dır. Bilinen
x fonksiyonu ile
r fonksiyonu ve böylece elektrostatik potansiyel
r , potansiyel enerji V r
, yoğunluk
r elde edilebilir.
Şekil 2.7. Thomas-Fermi denkleminin çözümü (Erkoç and Uzer, 1996)
Grafikte;
(1), Nötr atom çözümü; x ekseni için asimptotik olan bir çözüm.
(2), Pozitif iyon
N Z
için çözüm; xx0 sonlu bir değer için sıfır olan bir çözüm.(3), Basınç altında nötr bir atom için çözüm; x’ in büyük değerleri için ıraksayan ve asla sıfır olmayan bir çözüm.
Nötr atomlar için merkezi potansiyel V r
aşağıdaki gibi verilir;
2 0 4 Ze V r r (2.53) Burada
x 1 1, 588x...yazıldığında,
4 3 2 0 1, 794 Z Z V r e r a (2.54)sonucu elde edilir. Bu denklemin sağ tarafındaki ilk terim nükleer çekimi, ikinci terim ise elektron tepkisini temsil eder (Erkoç and Uzer, 1996). Denklem (2.54), SI birim sisteminde,
2 4 3 0 0 1, 794 4 e Z Z V r r a (2.55) şekliyle yazılabilir. 2.5.2. Hartree-Fock metoduHartree-Fock denklemi elektron probleminin çözümünde kullanılır. Born-Oppenheimer yaklaşımına dayanan modellerde, potansiyeli oluşturan
Radyabatik öz değerin çözümü için gereklidir. M çekirdek ve N elektronlardan oluşan moleküler bir sistem için elektronik Hamiltonien, elektron-elektron etkileşimlerinin ve tek elektronlu sistemlerin Hamiltonieni olan h(i)’ lerin toplamıdır. Yani
1 1 1 2 N i j ij H h i r
(2.56)
1 2 2 M i i Z h i r
(2.57)şeklinde verilir. Burada Z çekirdeğin elektrik yükü, ri riR ve rij ri rj
dir. Elektron dalga fonksiyonu , Schrödinger denklemini sağlar; n
1 1 1 2 N n n n i j ij h i r r r
(2.58)Denklem (2.58)’in çözümü için çeşitli yaklaşımları kullanmak gereklidir. Elektron probleminin çözümünde önemli yaklaşımlardan biri de Hartree-Fock yaklaşımıdır. Burada elektron dalga fonksiyonu, her bir elektronun dalga fonksiyonunun spin n orbitalleriyle çarpımlarından oluşur.
1
2
1 1 2 ... N k N k k N
(2.59)Bu orbitaller ortonormaldir. Yani
' '
k k kk
(2.60)
olarak yazılır. Elektronların antisimetrik özelliğini hesaba katmak için çarpım dalga fonksiyonları, antisimetrik bir ˆA operatörü tarafından tamamen simetrik hale
getirilir;
1 ˆ N k k A k
(2.61)Burada antisimetrik operatörü, ˆ 1
1 ˆ !p
A p
N
şeklinde tanımlanır. ˆp , birpermütasyon operatörüdür ve tüm elektron permütasyonlarının toplamıdır. ˆp
operatörünün ön kısmındaki işaret, çift bir permütasyon için pozitif, tek permütasyon için negatiftir. Sonuçta, Denklem (2.61) Slater determinantı olarak adlandırılan bir determinant şeklinde yazılabilir;
1 1 1 2 2 2 1 2 1 2 1 ! 1 2 A N N N N N N N , 1 det
1 2...
! A N N (2.62)HF yaklaşımında hamiltonienin beklenen değeri,
1 ˆ 1 1 2 N ij A A i ij ij p H i h i ij ij r
ifadesinden bulunur ve sonuçta1 1 2 N A A i ij ij i ij H h J K
(2.63)eşitliği yazılır. Burada 2. terim, Coulomb integralini,
2
2 2 1 12 1 1 1 2 ij i j ij J ij ij dx dx r r
(2.64) ve değiş-tokuş integralini,
* * 2 1 12 1 1 ˆ 1 2 2 1 ij ij i j i j ij K ij p ji dx dx r r
(2.65)içermektedir. Coulomb integrali Jij, klasik elektrostatik enerjidir fakat değiş-tokuş
integrali buna karşılık gelmemektedir (Jij Kij).
1 N i ij j j F
(2.66) şeklinde yazıldığında
1
1 HF
1F h v eşitliği ile F, Fock operatörü olarak
tanımlanır. vHF Hartree-Fock potansiyelidir ve
1 N HF i i i v j h j J j K j
(2.67)olarak ifade edilir. Ayrıca yukarıda verilen Fock operatörü hermityendir.
Denklem (2.66) kullanılarak orbital enerji,
i i ij ij
ij
i F i h J K
(2.68)şeklinde tanımlanır. Denklem (2.63)’de verilen beklenen değer bağıntısıyla HF yaklaşımında temel hal enerjisi,
1 1 2 1 2 N g i ij ij i ij N i ij ij i ij E h J K J K
(2.69)olarak verilir. Sonuç olarak HF yaklaşımında sıfırıncı mertebe Hamiltonien
0 i H
F i ve enerji 1 N o i iE
şeklinde yazılır. Denklem (2.69)’de temel durum enerjisi,0 1
g
olarak ifade edilir. Bu bağıntıda ilk sıra terimi ise
1 0 1 2 A A ij ij ij E H H J K
(2.71)HF yaklaşımı sınırlandırılmış Hartree-Fock (RHF) ve sınırlandırılmamış (UHF) olmak üzere iki farklı yaklaşım içermektedir. RHF yaklaşımında HF dalga fonksiyonu toplam elektron spinlerinin bir öz fonksiyonudur. Bu yaklaşım kapalı kabuk sistemler içindir. RHF yaklaşımına kıyasla UHF’nin kullanımı enerji değişimini genel olarak azaltabilir. Bu nedenle açık kabuklu sistemler için UHF, nümerik hesaplamalarda sıklıkla kullanılır. Ancak UHF yaklaşımının kullanımında HF dalga fonksiyonu toplam elektron spinin öz fonksiyonu değildir. Bu durum spin ilavesi olarak adlandırılır. Toplam elektron spininin öz fonksiyonunu elde etmek için farklı UHF dalga fonksiyonlarının lineer kombinasyonlarının biçimlendirilmesine ihtiyaç vardır (Zhang, 1999).
2.5.3. Hartree-SCF metodu
Self-Consistent Field (SCF) metodu, Hartree tarafından açıklanan bir metottur ve Öz Uyumlu Alan Metodu olarak bilinir. Atomik elektronların etkisini hesaplayan bu metot atomik potansiyelle birlikte enerji öz değeri ve dalga fonksiyonunu açıklar. Çok elektronlu atomun enerji ve dalga fonksiyonları da bu metotla nümerik olarak hesaplanabilir.
Atom numarası Z olan N elektronlu bir sistemde her bir elektron, bir diğer (N-1) tane elektronun yük yoğunluğuna göre merkezi bir potansiyelde hareket eder. Schrödinger denklemi, merkezi alanda her bir elektron için çözümlenir ve sonuçta elde edilen dalga fonksiyonunun daha önce hesaplanan dalga fonksiyonuyla uyumu incelenir.
Atomik dalga fonksiyonu, tek elektronlu ortonormal dalga fonksiyonlarının çarpımı ile verilir;
r r1, ,....2 rN
1
r1 2
r2 ....N
rN (2.72)
Moleküler dalga fonksiyonları ve enerjileri k E için yukarıdaki ifadelerin k matematiksel gösterimi;
ˆ k k k k k k H r E r (2.73)
2 2 2 k k k H W r m (2.74)
k c
k k
k W r V r V r (2.75)şeklindedir. Burada V rc
k elektron-elektron Coulomb enerjisi, V rk
k ifadesi de2
jk
e r
terimidir. Bu denklemler iterasyon yapılarak çözülür. Merkezi yaklaşım
potansiyeli için W r
k deneme fonksiyonu olarak tahmin edilir ve burada W 1
r kile gösterilir;
1
k k
W r W r (2.76)
Elektronun dalga fonksiyonları bu yaklaşım potansiyelinde;
2 1 1 1 2 2m k W rk k rk Ek (2.77)ifadesiyle hesaplanır. Birinci mertebede dalga fonksiyonları olan k 1
rk ile yük yoğunluğu e2k 1
rk 2 hesaplanır. Bir önceki sonuçlar yardımıyla ikinci mertebe atomik potansiyel için de k 2
rk hesaplanır.Bu işlem adımları başlangıçtaki değer sonuçta elde edilene kadar yani
1
n n
k k
W r W r sonucuna kadar devam edilir. Bu sonuçlar içerisinde en küçük enerjiye karşılık gelen dalga fonksiyonu öz fonksiyondur. Başlangıçta kabul edilen değer ile sonuçta elde edilen değerin uyumundan dolayı bu metoda Öz Uyumlu Alan Metodu denir.
Helyum atomu için Hartree-Fock metodunu uygulamada iki elektron dalga fonksiyonu,
r r1, 2
r1 r2 (2.78)
şeklinde yazılır. Denklemde 2. elektronun olasılık dağılımı, *
r1 r dr2 2 şeklinde yazılır. Böylece 1. elektronun 2. elektron nedeniyle r noktasında sahip olduğu etkin 1potansiyel enerji,
1
N etkin i i j j j j i ij V r j r r
(2.79)
2 *
1 1 2 2 2 12 1 etkin V r e r r dr r
(2.80)şeklinde yazılır. Burada etkin Hamiltonien operatörü,







![Çizelge 4.11. BzChCl’nin konformer yapılarını tanımlayan torsiyon açıları Torsiyon Açıları( 0 ) Deneysel* Konformer I Konformer II Konformer III Konformer IV 1 [C(21)-O(20)-C(1)-C(4)] 167,9 115,02 83,64 -179,64 136,13 2 [O(20)-C(1)-C(4)-](https://thumb-eu.123doks.com/thumbv2/9libnet/4025560.55858/89.892.173.789.211.440/yapılarını-tanımlayan-açıları-açıları-konformer-konformer-konformer-konformer.webp)

