^ 'i ·. . î ' * ^ : ' w ¿ · ^ . i ; .,■· i v іГ Γ ί ΐ , Ζ < . ■■^-· ■ ·-· , ' ^ >· . <1 " ·ΐ ‘^ · * · < · > ¡ » - '« • « ϋ · ■« l U . я *·· * ► ·
; !·Η
АСТ'ѵ'ІТ;-·!;
''"3 Tv.3':OR
ΡίίΟ'ΓΞίί·!
А Û! Ζ ^'‘· * 'Cf С ί ’ ■■ ; I A i гіі:і ^ \ L i .
IDENTIFICATION OF THE ROLE OF PROTEIN IN THE CELLULAR ACTIVITIES OF p53 TUMOR SUPPRESSOR PROTEIN
A THESIS SUBMITTED TO
THE DEPARTMENT OF MOLECULAR BIOLOGY AND GENETICS AND
THE INSTITUTE OF ENGINEERING AND SCIENCE OF BiLKENT UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE
By
N.C.TOLGA EMRE July, 1999
6»P tiL ,:-/? · ■Р-/Ц
t L|fe
I certify that I read this thesis and that in my opinion it is fully adequate, in scope and in quality, as thesis for the degree of Master of Science.
I^rof. Df. Mehmet
I certify that I read this thesis and that in my opinion it is fully adequate, in scope and in quality, as thesis for the degree o f Master of Science.
Prof. Dr. Meral Özgüç
I certify that I read this thesis and that in my opinion it is fully adequate, in scope and in quality, as thesis for the degree of Master of Science.
AssisK Prof. Rengiil Çetin-Atalay
Approved for Institute of Engineering and Science.
Prof.Dr. Mehmet Bafay, Director of Institute of Engineering and Science
ABSTRACT
IDENTIFICATION OF THE ROLE OF PROTEIN IN THE CELLULAR ACTIVITIES OF p53 TUMOR SUPPRESSOR PROTEIN
N.C.TOLGA EMRE
M.S. in Molecular Biology and Genetics Supervisor: Assist. Prof. Rengiil Cetin-Atalay
July 1999, 96 Pages
p53 is a tumor suppressor gene which is mutated in about 50% of human cancers. The product of p53 gene encodes a sequence-specific transcription factor. The genes transactivated by p53 code for proteins that are implicated in the negative regulation of cell proliferation (via apoptosis or cell cycle arrest) and DNA damage repair. p53 protein interacts with several viral and cellular proteins and these interactions are important in the regulation and dysrégulation of the functions of p53. Another gene, named INGl (for “Inhibitor of Growth 1”), was identified as a candidate tumor suppressor gene due to its functions in apoptosis and cell cycle arrest. P33ING1 protein product of INGl, was shown to enhance the growth suppressive functions of p53. Furthermore, a physical association between p53 and p33^*^°* proteins has been detected by immunoprécipitation. In this study, we investigated the physical interaction between p53 and p33^’^°‘ using in
vitro methods in order to determine the region of p53 that enabled this
interaction. As a preliminary step for the study, the INGl cDNA was amplified from total cDNA of a cell line by PCR and cloned into expression vectors. The recombinant p33^^*^’ protein was overexpressed in E.coli and purified as a fusion protein with GST. The wild-type p53 cDNA and its several deletion mutant constructs were subcloned into a suitable expression vector to enable subsequent in vitro transcription-translation reactions. 1«
vitro transcription-translation products of these constructs were used in GST
pulldown assays with purified GSTp33 protein to map the interacting region on the human p53 protein. The results of the study suggest that the primary determinant on p53 protein in its interaction with p33^’^°\ is the C-terminal domain while there may be other regions that are involved in the interaction.
ÖZET
p33iNGi PROTEİNİNİN p53 TÜMÖR BASKILIYiCI PROTEİNİNİN HÜCRESEL AKTİVİTİLERİ ÜZERİNDEKİ ETKİLERİNİN BELİRLENMESİ
N.C. TOLGA EMRE
Yüksek Lisans Tezi, Moleküler Biyoloji ve Genetik Tez Yöneticisi: Yard. Doç. Dr. Rengül Çetin-Atalay
Temmuz 1999, 96 Sayfa
p53, insan kanserlerinin yaklaşik %50’sinde mutasyona uğrayan bir tümör baskılıyla gendir. p53 gen ürünü, DNA dizisi özgünlüğü olan bir transkripsiyon faktörüdür. p53 tarafından transkripsiyonu aktive edilen genlerin protein ürünleri, hücre bölünmesini (apoptoz veya hücre döngüsün bloku yolu ile) önleyici yönde görev yapan kontrol elemanlar! ya da DNA hasarı onarım sistemi elemanlarıdır. p53 proteini birçok hücresel ve viral protein ile etkileşime girer ve bu etkileşimler p53’ün işlevlerinin düzenlenmesinde veya düzensizliğinde önemli yer tutar. INGI (“Inhibitor of Growth 1”: Büyüme İnhibitörü 1) adli yeni bir gen , apoptoz ve hücre döngüsü blokundaki rolleri dolayısıyla tümör baskılayıcı gen adayı olarak tanımlanmıştır. IN G l’in protein ürünü olan p33^^°^’in, p53’ün büyüme baskılıyla işlevlerini arttırdığı gözlemlenmiştir. Dahası, p53 ile p33™°^ arasında fiziksel bir etkileşim, immunopresipitasyon yöntemiyle belirlenmiştir. Bu çalışmada, p53’ün p33^’^°' ile etkileşen bölgesini belirlemek amacıyla bu iki protein arasindaki fiziksel etkileşimi in vitro yöntemlerle inceledik. İlk adım olarak INGI cDNA’sı, bir hücre dizisi toplam cDNA’sından polimeraz zincir tepkimesi yöntemiyle çoğaltıldı ve ekspresyon vektörlerine klonlandı. Rekombinant p33’'’^°* proteini GST füzyon proteini olarak E.coIVğq üretildi ve saflaştırıldı. Normal p53 cDNA’sı ve birçok değişik p53 delesyon mutantı cDNA’ları, in vitro transkripsiyon-translasyon tepkimelerinin yapilmasina elverişli vektörlere subklonlandı. Daha sonra, bu tepkime ürünleri, saflaştırılmış GSTp33 proteini ile birlikte p53’ün p33™°* ile etkileşen bölgesini belirlemek amaciyla GST-indirme sisteminde kullanıldı. Çalişmanin sonuçları öyle göstermektedir ki p33™°* ile etkileşmede p53 proteini üzerindeki ana etken, C-terminal bölgesidir, ancak diğer bölgelerin de katkısı olabilir.
Anahtar Kelimeler. p33™°’, p53, tümör baskılıyla, protein-protein etkileşimi,
ACKNOWLEDGEMENT
Thank you...
...Dr. Rengül Çetin-Atalay, my supervisor, for all that I have learned from you, for your warmth and patience.
...Dr. Mehmet Öztürk, for the knowledge and experience you have shared and the support you have given all through these two years.
...My family, including Grandmom, because you have always been here, in Bilkent, with me.
...Ash, hardest to adapt, and hard to give up, and Gülayşe, the sweet smile; you made tough days easier.
...My home mates. Tolga, for the friendship and support in the lab, in kitchen, and in the thesis; and Bleda, though things should have worked out to be better.
.. .Friends in Istanbul, especially Burak and Tayfun, kilometers between did not exhaust your support and Başar, old friend a new neighborhood, I wished I had the chance to see you more often.
...Hani, my neighbor for a year, for all the times we pretended together. ...Esma, Hilal the ancient Goddess in vein, and Arzu the “komşu”, for not withholding what you know.
...Dr. Ergün Pınarbaşı, Dr. Uğur Yavuzer and Dr. Işık Yuluğ, for the help, intellectual and material.
...Birsen, for the DNA sequencing.
...The labmates, past and present, especially Çağla, Buket, Cemaliye, Tuba and Emre, but also Reşat, Korkut, Berna, Alper, Gökçe, Necati, Abdullah and Nuri, for the help or the small talks.
...Our support units, Abdullah Bey,Sevim Hanım, Tülay and others, for the efforts you have spent to make things run smoothly.
TABLE OF CONTENTS
SIGNATURE PAGE... ii ABSTRACT... iii ÖZET... iv ACKNOWLEDGEMENT...v TABLE OF CONTENTS...vi LIST OF FIGURES...x LIST OF TABLÉS...xii ABBREVIATIONS...xiii CHAPTER 1. INTRODUCTION...2 1.1 General Introduction...21.2 p53 Tumor Suppressor Gene and Protein Product...3
1.2.1 Background... 3
1.2.2 The p53 gene... 5
1.2.3 Cellular Functions of the p53 protein... 5
1.2.4 Regulation of p53 Activity...7
1.2.5 Downstream Events...10
1.2.6 p53 Mutations... 13
1.2.7 Structure of the p53 protein...15
1.2.8 Interaction of p53 Protein with Other Proteins...19
1.3 INGl Gene and its Protein Product, p33^^°*... 19
2.1 MATERIALS...23 2.1.1 Chemicals... 23 2.1.2 Bacterial Strains... 23 2.1.3 Enzymes... 23 2.1.4 Oligonucleotides... 24 2.1.5 Cloning Vectors... 24 2.1.6 Antibodies... 25
2.1.7 Commercially Available Kits... 26
2.1.8 Apparatus... 26
2.1.9 Materials for Protein Purification...27
2.1.10 Materials for Autoradiograghy... 27
2.1.11 DNA and Protein Size Markers...28
2.1.12 Tissue Culture Reagents and Apparatus...28
2.2 SOLUTIONS AND MEDIA...29
2.2.1 Agarose Gel Electrophoresis Solutions... 29
2.2.2 Solutions for Plasmid DNA Isolation (MiniPrep)...29
2.2.3 Solutions for Bacterial Transformation... 29
2.2.4 Microbiological media and antibiotics...30
2.2.5 Polyacrylamide Gel Electrophoresis Solutions... 30
2.2.6 Protein Purification Buffers... 31
2.2.7 Solutions for Protein Concentration Estimation...31
2.2.8 Western Blotting Solutions... 31
2.3.1 PCR amplification of cDNA... 33
2.3.2 Agarose gel electrophoresis of DNA...33
2.3.3 Recovery of DNA Fragments from Agarose Gels... 34
2.3.4 Methods for Cloning and Subcloning of Genes... 34
2.3.4.1 Restriction Enzyme Digestion of DNA...34
2.3.4.2 DNA Ligation Reactions... 35
2.3.4.3 Transformation o iE .co li... 35
2.3.4.4 Plasmid DNA Preparation... 36
2.3.5 Quantification and Qualification of Nucleic Acids... 38
2.3.6 Growth and Storage of Bacterial Strains... 38
2.3.7 SDS Polyacrylamide Gel Electrophoresis...38
2.3.8 Protein Expression and Purification...39
2.3.8.1 Cell Growth and Induction... 39
2.3.8.2 Cell Harvest and Lysis... 40
2.3.8.3 Protein Purification with Glutathione-Bound Resins... 41
2.3.8.4 Concentrating the Purified Proteins... 42
2.3.8.5 Quantification of Purified Proteins...42
2.3.9 Immunological Detection of Immobilized Proteins(Western Blot) 43 2.3.10 In vitro Transcription-Translation (IVTT) Reactions... 44
2.3.11 GST Pull-Down Assays...45
2.3.12 Polyclonal Antibody Production... 47
2.3.13 Tissue Culture Techniques... 48
2.3.13.1 Cell Line Maintenance and Propagation...48
2.3.13.2 Cell Lysis for Western Blotting... 48
2.3.14 Automated DNA Sequencing... 49
CHAPTER 3: RESULTS... 50
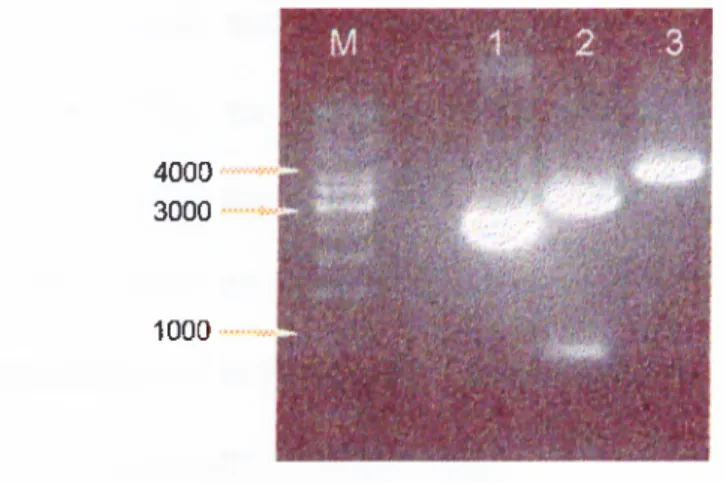
3.1 PCR Amplification of p33^^*^^ cDNA...50
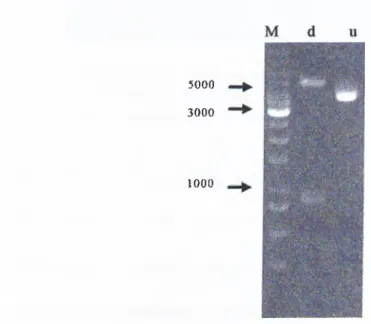
3.2 Construction of the pBSK-p33™°* Recombinant Phagemid...52
3.3 Subcloning of p33™°* cDNA... 54
3.4 Small-scale Induction of pGEXp33 Containing Clones... 56
3.5 Large-Scale Expression and Purification of GST-p33 Fusion Protein 57 3.6 Subcloning of p53 cDNA and p53 Deletion Mutant Constructs...58
3.7 Polyclonal Rabbit anti-GSTp33 Antibodies... 61
3.7.1 Antibody Production and Characterization... 61
3.7.2 Clearing of anti-GST antibodies...62
3.8 Preliminary Analysis of p33™*^* Expression In Cell Lines... 63
3.9 IVTT of p53 and its Deletion Mutants... 65
3.10 Mapping the Interaction Region of p53 with p33™*^^ by GST Pulldown66 CHAPTER 4: DISCUSSION... 72
REFERENCES... 78
APPENDICES...87
Appendix A: GENBANK Report of p33^^*^' cDNA and protein product... 88
Appendix B; GENBANK Report of INGl-L cDNA and conceptually translated protein product... 90
Appendix C: Alignment of p33**^°‘ and INGl-L protein sequences... 92
Appendix D: Characteristics of MCF-7 cell line... 93
LIST OF FIGURES Figure 1.1 Figure 1.2 Figure 1.3 Figure 1.4 Figure 2.1 Figure 2.2 Figure 3.1 Figure 3.2 Figure 3.3 Figure 3.4 Figure 3.5 Figure 3.6 Figure 3.7 Figure 3.8 Figure 3.9
Summary of upstream and downstream events in p53-
mediated cellular responses...6
Summary of posttranscriptional modifications and domains of p53... 9
Major p53-dependent pathways...13
Domain structure of p53, together with mutational hotspots...17
Vector map of pBluescript II KS'... 25
Migration patterns of size markers... 28
AGE of p33INGl PCR-products... 51
AGE analysis of MidiPrep of pBskp33 construct...53
AGE of restriction enzyme digestion analysis of pGEXp33 construct... 55
Small-scale induction of selected pGEXp33 containing clones...56
Analysis of purified GSTp33 protein...57
Restriction enzyme digestion analysis of MidiPreps of deletion mutant constructs... 60
Characterization of polyclonal anti-GSTp33 antibody with Western Blotting...63
Western Blotting of cell line lysates with polyclonal rabbit anti-p33 antibodies... 64
SDS-PAGE analysis of IVTT reactions of deletion mutant constructs...65
Figure 3.10 Figure 3.11 Figure 3.12
GST Pulldown Assay of wt p53 with GSTp33...66
GST pulldown with p53 deletion constructs...68
LIST OF TABLES
Table 1.1 Table 1.2
Tables. 1 Table 3.2
Major genes transactivated by p53... 11 Representative examples of proteins interacting with p53... 18
Deletion mutant constructs of p53... 59 Summary of GST Pulldown Assay Results...69
ABBREVIATIONS
Aeoo absorbance at 600 nanometer wavelenght AGE agarose gel electrophoresis
APS ammonium persulfate bp base pair
BSA bovine serum albumin cDNA complementary DNA
cm centimeter
ddHjO deionized, distilled water
DMEM Dulbecco's Modified Eagle Medium DNA deoxyribonucleic acid
dNTP deoxyhucleotide triphosphate EDTA ethylenediaminetetra-acetic acid FGS fetal calf serum
g gram
hr hour
HRP horse reddish peroxidase
IPTG isopropylthio-beta-D-galactoside IVTT in vitro transcription and translation
kb kilobase kD kilodalton LB Luria-Bertani medium M molar ME mercaptoethanol min minute
MidiPrep medium-scale isolation of plasmid DNA by alkaline lysis method MiniPrep small-scale isolation of plasmid DNA by alkaline lysis method ml milliliter
mRNA messenger RNA
nm nanometer
NT A nitrilo-tri-acetic-acid
PBS phosphate buffered saline
pBsk pBluescript II KS' phagemid vector PCR polymerase chain reaction
PVDF polyvinilydene difluoride rpm revolution per minute RNA ribonucleic acid RNase ribonucléase
SDS sodium dodecyl sulphate
sec second
TAE tris/acetic acid/EDTA buffer
TEMED N,N,N,N-tetramethyl-1,2 diaminoethane Tris 2-amino-2-[hydroxymethyl]- 1,3 propandiol
U unit
V volt
v/v volume for volume
wt wild type
w/v weight for volume w/w weight for weight
CHAPTER 1. INTRODUCTION
1.1 General Introduction
Cancer is a disease of uncontrolled proliferation of cells and cancer development is a result of multiple events. Normal cells of multicellular organisms have several mechanisms to regulate their growth and differentiation, therefore, various different alterations may be required to bypass these controls. Most of these alterations involve either the activation of oncogenes or inactivation of tumor suppressor genes.
Oncogenes were initially identified as genes carried by viruses that cause transformation of their target cells. Their cellular homologues (also called proto-oncogenes) were found to be genes with normal functions in the cellular physiology (e.g. ras, myc), but a mutation or dysrégulation in expression leading to pathogenic overactivity of these genes resulted in uncontrolled cellular proliferation and tumor formation. These alterations result in a gain^of-function: a single allele of the mutant gene which is dominant over its normal allelic counterpart, is sufficient to contribute to carcinogenesis.
However, studies in somatic cell genetics suggested yet another class of genes whose products seemed to inhibit neoplastic transformation, introducing single chromosomes or certain chromosome parts inhibited the capacity of cultured, transformed cells to induce tumors in animals, therefore, this class of
genes were named tumor suppressor genes. The products of the tumor suppressor genes are required for normal cell function, and they antagonize the proliferative actions of oncogenes to prevent tumorigenesis. Contrary to oncogenes, defects in both copies of a tumor suppressor is required to abolish its functions. So tumor suppressor gene mutations act as a recessive trait in the formation of tumors. This recessive nature of tumor suppressor genes results in familial cancer syndromes where one non-functional copy of the gene is transmitted through generations (e.g. Li-Fraumeni syndrome as a result of germline p53 mutations). Tumor formation in the offspring is observed when the other, functional allele is inactivated by a somatic mutation (e.g. deletion) during the course of the life. This situation is known as “Loss of heterozygosity (LOH)”, since the initial heterozygous genotype of the individual is abolished by the loss of thè functional âllélè.
Tumor suppressor genes constitute a very heterologous class of genes in terms of functions of their protein products. They can be transcription factors (e.g. p53, WT-1), cyclin dependent kinase inhibitors (e.g. p l6), GTPase activator molecules (e.g. NFl), or receptor tyrosine kinases (e.g. RET)(for reviews, see Levine, 1993 ; Klein, 1997 ) .
1.2 p53 Tumor Suppressor Gene and Protein Product 1.2.1 Background
One of the main subjects of current cancer research is p53, a tumor suppressor gene and its protein product. This interest is primarily due to the observation that more than 50% of human tumors have mutations in the p53
gene. Furthermore, p53 is the key element in negative regulation of cell proliferation which is a means for preventing neoplastic transformations (for reviews, see Wang & Harris, 1997 ; Ozbun, 1997 ; Adams & Kaelin, 1998 ; Almog & Rotter, 1998 ; Ko & Prives, 1996 ).
p53 was identified in the late 1970s as a cellular protein which was bound to the viral oncoprotein, large tumor antigen (T-ag), in cells transformed by simian virus 40 (SV40).(Lane et al., 1979; Linzer et al., 1979) In the beginning of 1980s, the murine p53 gene was cloned and sequenced. Early studies suggested that p53 was capable of immortalizing cells and cooperating with an activated ras oncogene to transform primary fibroblasts (Parada et al., 1984). These studies indicated that p53 should be categorized as a cellular oncogene (Jenkins et al., 1984) However, subsequent studies revealed that p53 cDNAs used in these studies were mutant and wild-type counterparts were shown to suppress the growth of transformed cells in vitro and tumorigenic potential of cells in animals (Finlay
et al., 1989) . Mutations of p53 alleles were implicated in many human and
animal tumors, and Li-Fraumeni syndrome, a familial cancer susceptibility syndrome, is associated with germ-line transmission of a mutated p53 allele (Malkin et al., 1990) The role of p53 in cancer was more directly accessed by the observation that p53 knock out mice developed tumors at high frequencies (Donehower et al., 1992). These observations indicated that p53 should be re-classified as a tumor suppressor gene.
1.2.2 The p53 gene
The human p53 gene resides on the short arm of chromosome 17 (i.e., 17pl3.1). Many organisms have p53 homologues (such as monkey, chicken, rat, frog) while some commonly investigated model animals do not seem to have homologues (such as fruit fly, sea urchin, yeast) (Soussi & May, 1996) . The gene, spanning 12 kb of genomic DNA, has 11 exons with 1*' exon non coding. Two promoter regions, both lacking consensus TATA or CAAT boxes, have been implicated in the regulation of the gene. A positive regulatory element is located between exons 4 and 5 (Ozbun et a l, 1995) . Expression levels of the p53 gene seems to correlate with the proliferative state of cells. Highest expression levels are observed just before the entry to S-phase of cell cycle.
1.2.3 Cellular Functions of the p53 protein
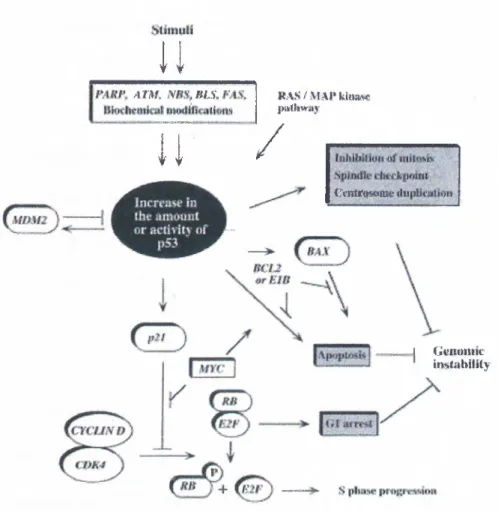
The tumor suppressor activity of p53 is a result of its ability to act as a negative regulator of cell proliferation. Such regulation is achieved by either a cell cycle arrest or induction of programmed cell death (i.e., apoptosis). The molecular pathways leading to either of such consequences is mainly due to the fact that p53 protein is a sequence-specific transcription factor that can transactivate or repress a variety of target genes that are responsible for eliciting the required responses (for reviews, see Giaccia & Kastan, 1998 ; Agarwal et al., 1998 ; Hansen & Oren, 1997 ).
In order to prevent neoplastic transformations, p53 aims to maintain genomic stability whose loss can lead to unwarranted overactivity of oncogenes or mutations in other tumor suppressor genes leading
tumorigenesis (Yin el ah, 1992 ). For this reason, p53 is said to be “the guardian of the genome”. Several genotoxic stress situations are known to activate p53 (Figure 1.1) ; DNA damage (due to radiation or chemical mutagens)(Kastan et a l, 1991), hypoxia (Graeber et al., 1994), redox stress (Hainaut et al., 1993) and activated oncogenes (Serrano et al., 1997). The state of a cells adhesion (Nigro et al., 1997) and depletion of nucleic acid reservoirs (Linke et al., 1996) are also implicated in the activation of p53, but mechanisms involved are yet largely unknown.
IRAJV Hypoxia REDOX
Alteration Oncogene Activation Adhession rNTP Denletion Cell cycle arrest
(or other accessory nrolcins'l
Apoptosis
Phosphorylation/ Phosphorylation/ Phosphorylation/ p l 9 ' ^ Integrin? RNA Signalling? A cetylation HIFl binding R cfl interaction
1.2.4 Regulation of p53 Activity
In normal cells, levels of p53 molecules are very low due to its short half-life. It is only when there is a stress condition that may lead to genomic instability, the presence of p53 protein is required. This requirement is fulfilled by the rapid stabilization and activation of the p53 molecules. Activation of p53 is mainly posttranslational. This property enables a fast response to genotoxic stresses when compared to activation at the transcriptional level. As a first step in p53 activation, p53 protein is stabilized to increase its originally short half life. Additional steps are required to activate the stabilized proteins (Chernov et a l, 1998 ). Several different modifications are implicated in the activation of the p53 protein, such as phosphorylations, déphosphorylations, acetylations, and binding to certain proteins. There are several serine residues in both N- and C-terminal domain of the protein which are targets for phosphorylation by various kinases (e.g., ATM, DNA-PK, CKI, MAP kinase). Certain physiological stimuli results in the phosphorylation or dephosphorylation of certain residues, for example ATM protein is shown to phosphorylate ser-15 in response to ionising radiation (IR) while ser-376 dephosphorylation is also associated with IR- induced DNA damage, which, in turn, enables 14-3-3 protein binding to p53, leading to G1 arrest (Waterman et a l, 1998 ).
Upon UV-induced DNA damage, acetylation of lysine residues in the N-terminal domain by p300/CBP and PCAF histone acetylase family proteins cause an increase in the sequence-specific DNA binding capacity of p53 (Lill
é ta l, 1997).
Activation of p53 protein can also be facilitated by binding to cellular proteins. Examples in this category are Refl, a redox/repair protein (Jayaraman et a l, 1997) and HMGl, a protein involved in DNA bending that enhances the tetramer formation of p53 molecules on the target DNA sequences (Jayaraman et a l, 1998). As seen in the examples, regulation of the activation of latent p53 molecules is a complex process, however, considering the variety of stress conditions upstream of p53 and various downstream responses (Figure 1.2), this tight regulation should not be surprising. The common denominators in the activation process can be categorized as increasing the half-life of p53 protein by inhibiting its interaction with Mdm2 and increasing the sequence specific DNA binding ability by relieving the C- terminal block.
Translational control, in which initiation of translation from p53 mRNAs is increased, is also implicated in the induction of p53 (Mosner et a l, 1995). Regulation at the level of transcription seems to be less prominent.
Direct inactivation of p53 protein is achieved by binding of p53 to Mdm2, a protein whose overactivity was shown to be tumorigenic. Mdm2 binding inhibits the transactivation by p53, and further, targets p53 for degradation by the ubiquitine pathway (Haupt et a l, 1997). Mdm2 expression is transactivated by the p53 protein, thus, Mdm2 and p53 constitute an autoregulatory feedback system in which p53 limits its own activity (Wu et
u v IR V PK C /PP.«.V \ Ш
IR \ \ \
TAP 14-3^ В«№Мб ■AuosTprac” COHTRDlRESieMNEGATIVE TUMOR MUIATIQI^
REGULATCKRY SV40TAINDII№
DOMAIK
QUGOMERIZATIOK
Figure 1.2 Summary of posttranscriptional modifications and domains of p53. From G iaccia &Kastan, 1998.
It has been known for some time that overactivity of oncogenes can lead to p53-mediated cell cycle arrest or apoptosis (for example, see Hermeking ei
a l, 1994). Recently, a series of experiments shed light to the mechanisms
leading to these responses. A new protein, denoted ARF (Alternative Reading Frame; pl9 in mice, pl4 in humans), has been shown to be expressed by using a novel reading frame from the INK4 locus (Kamijo ei al., 1997). INK4 locus has been known to code for p i6, a cyclin-dependent kinase inhibitor that acts as a tumor suppressor. ARF protein can bind p53 and Mdm2 proteins and this binding facilitates p53 stabilization by relieving of p53 from Mdm2 sequestration (Kamijo ei al., 1998). Мус overactivity was shown to increase ARF levels through direct transactivation of the ARF expression. In the absence of oncogene overactivity, p53 protein can transrepress the ARF promoter, forming a negative feedback loop similar to the case of Mdm2 (for a review, see Prives, 1998). These autoregulatory loops ensure that p53 level in the cell is low unless there is a stress condition.
1.2.5 Downstream Events
p53 interacts with regulatory sequences of several genes leading to either activation or repression of their expression (Table 1.1). Downstream targets activated by p53 are components of DNA recombination/repair system, apoptotic machinery or cell cycle regulators (Figure 1.3).
p53-dependent cell cycle arrest is mainly imposed at the Gi-phase by the transactivation of (el-Deiry et al., 1993), a member of the small cyclin-dependent kinase (CDK) inhibitor (CKI) family. inhibits the phosphorylation activity of the cyclinE- cdc2 complex which ultimately results in the blocking of transition to S-phase (Slebos et al., 1994). p2i''''^^^^**'* is also known to inhibit proliferating cell nuclear antigen (PCNA), displacing DNA polymerase-delta, thus blocking replication (Waga et al., 1994). A similar effect on PCNA is observed with another p53-responsive gene, GADD45 . Cell cycle arrest at G2 stage of cell cycle is also mediated by p53 in some situations (for example, see Stewart et al., 1995). Additionally , p53 deficient fibroblasts were shown not to respond to spindle inhibitors and go multiple rounds of replication without proper chromosome segregation, thus leading to aneuploidy, suggesting a role for p53 in spindle checkpoint (Cross
p21AVAFl/Cipl CDK inhibitor, binds PCNA (cell cycle arrest)
(el-Deiry, 1993 ) Mdm2 oncoprotein, p53 inhibitor (Momand, 1992), (Wu, 1993 ) GADD45 DNA repair; cell cycle arrest
through PCNA binding
(Kastan, 1992 ) Bax antiapoptotic; bcl2 family (Miyashita, 1995 ) IGF-BP3 insulin-like growth factor
binding protein-3; blocks signalling of a mitogenic growth factor
(Buckbinder, 1995 )
Table 1.1 Major genes transactivated by p53. Modified from Levine. 1997.
p53 influences replication by binding to replication protein A (RPA) leading to the inhibition of initiation of replication. This serves as a mechanism to delay DNA replication in case of DNA damage, since replication in the presence of damaged DNA endangers the integrity of the genome (Dutta et al., 1993). p53 is also implicated in recombination repair through its capability to bind damaged (single stranded) DNA and through its interaction with RAD51 protein (Buchhop et al., 1997 ).
Apoptosis is one of the major mechanisms whereby p53 can prevent tumorigenesis by eliminating cells that are in a pre-neoplastic stage (e.g. having DNA damage that is not repairable, see Evan , 1998 ). p53 may employ a transactivation-dependent pathway or direct protein-protein interactions to induce apoptosis (Haupt et al., 1995 ). A well-known mechanism involves the transactivation of bax, a tumor suppressor gene whose protein product interacts with bcl-2 protein (Miyashita et al., 1995 ). bcl-2 is an anti- apoptotic (survival) factor when it exists as homodimers. Increased bax expression leads to heterodimerization with bcl-2 which causes apoptosis.
Whether cell cycle arrest or apoptosis will take place as a result of p53 activation is dependent on the cell type, extent of DNA damage and the presence of extracellular growth and survival factors .
Under normal conditions, p53 protein is not essential for cellular maintenance or embryonic development. This is manifested in the observations that p53 knock out mice has normal development (Donehower et ah, 1992), except probably in haemopoetic cell lineages where overexpression of p53 induces the more differentiated state (Johnson et a i, 1993 ) .
Many years after the discovery of p53, two homologues of this gene, named p73 (Kaghad et al., 1997) and KET (Schmale et al., 1997 ) were identified ( for review, see Kaelin , 1999 ). p73 gene product is shown to tetramerize, transactivate p53 target genes, perform cell cycle arrest and induce apoptosis (Kaghad et al., 1997; lost et al., 1997 ). Despite these similarities with p53, it is not yet settled whether or not p73 is a functional homologue of p53: p73 is not induced upon DNA damage and p73 is not a target of viral protein-mediated degradation. Furthermore, p73 null mice do not develop spontaneous tumors but there is 60% lethality during the first 3 weeks (Kaghad et al., 1998). Unlike p53, which is ubiquitously expressed, both KET and p73 show tissue specific expression. Both genes have alternatively spliced variants. These observations suggest that although homologous, these proteins may have different functional roles in the cell.
u
/tm iBLl KA8VMAI*ymMj ¡»kdlWiiyI T
/
iVntri^ttWir , ,J G i'iiM iiiic iiiiStiibliilyFigure 1.3 Major p53-dependent pathways. From Agarwal et al., 1998 .
1.2.6 p53 Mutations
Mutations in the p53 gene are the most commonly detected gene alterations in human cancers (for review, see Greenblatt et al., 1994). Frequency of p53 mutations differ among types of tumor, ranging from 20% to 80% with an overall average of about 50%. Although all types of mutations (deletion, insertion, chain termination) are observed, the most common alterations are single base pair substitutions leading to single amino acid substitutions (“missense” mutations). Most of the p53 mutations associated with human neoplasia are observed in the core sequence-specific DNA binding domain of the protein. Furthermore, these mutations are localized to four of
the five evolutionarily conserved regions (regions II-V) and mutation hotspots are generally arginine residues (R175, R248, R249 ,R273 ,R282) (for reviews, see Greenblatt et al.\ 1994 ,Soussi and May, 1996). These arginines are critical in maintaining the overall conformation of the protein and its contact with target DNA (Cho et al., 1994 ) . Interestingly, some enviromental risk factors are shown to be associated with certain kinds of mutations, as in the case of the correlation of high dietary intake of aflatoxin with codon 249 mutations (Ozturk, 1991). It is important to note that some mutant proteins do not exhibit only a non-functional p53 phenotype; but they may also contribute to tumorigenesis by a dominant-negative effect (i.e., mutant proteins heterodimerizing with the wild-type proteins to render them inactive) and gain-of-function (i.e., by transactivation of genes that are actively promoting neoplastic transformation) (Dittmer ei al., 1993 ).
Mutations are not the only means to provide a p53-null phenotype; functional inactivation of the protein is also implicated in several tumor cases. Such inactivation can be achieved by increasing the rate of p53 degradation, for example by overexpression of Mdm2 protein or by binding of certain tumor virus proteins, such as E6 protein of HPV or ElB protein of adenovirus. Another possible way to block p53 activity is to modulate its subcellular localization: inability of p53 protein to reside in the nucleus prevents its transactivation functions. For example, in neuroblastoma tumors, p53 is not mutated, but it is found mostly in the cytoplasm. This phenomenon may be related to the NLS (nuclear localization signals) and/or NES (nuclear export signals) found in the protein, as described below(Stommel et al. 1999).
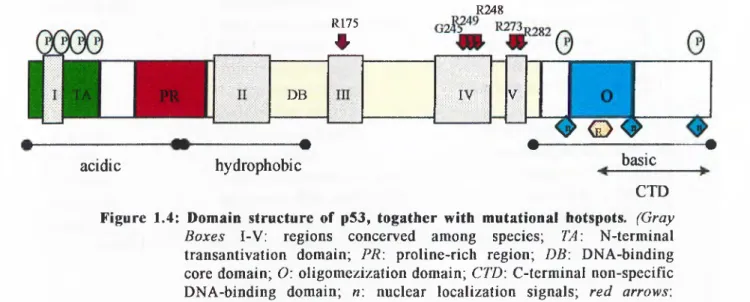
1.2.7 Structure of the p53 protein
Human p53 protein is a single polypeptide of 393 amino acids. Five regions in the protein, denoted I-V, are conserved among many diverse species (Soussi & May, 1996). The functionally active form of the protein which acts as a transcription factor, is a tetramer.
p53 protein can functionally and structurally be divided into five domains (Figure 1.4) (for reviews, see Prives, 1994 ; Levine, 1997 ):
- N-terminal transcriptional activation domain spans amino acid residues 1 to 43. It has mainly acidic residues and displays similarities to acidic activator domains of other transcription factors (such as VP 16). Among others, N-terminal domain binds TAFn40 & 60 through which p53 exerts its transcriptional activation role on target gene (Thut et al., 1995). Binding of the same domain to TBP is thought to be responsible for the transcriptional repression by p53 (Seto et al., 1992 ). Mdm2, the repressor of p53, also binds to this domain to inhibit p53’s transcriptional activation functions. Most of the phosphorylation sites of p53 protein also reside in this domain. Although single mutations occurring here does not abolish p53 transactivation functions, double mutations of certain critical residues do (Lin et al., 1994). This implies that overall 3-D structure of the domain is the primary determinant of the domain’s function.
- Proline rich region spans the amino acids 63 to 97. There are 5 tandemly repeated proline rich motifs in the form of PXXP (where P is proline and X is any amino acid) and these motifs were shown to bind SH3-containing
proteins. This region is implicated in both growth arrest and apoptosis (Venot et al., 1998 ). Additionally, proline rich region is also shown to contribute to the negative regulation of p53 functions, possibly in cooperation with the C-terminal domain, through an unknown mechanism (Muller-Tiemann
et al., 1998 ).
- DNA binding (core) domain spans residues 102 to 292. This is an independently folding unit which is resistant to cleavage by proteases, implying that the region is not accessible to solvent. Four of the five evolutionarily conserved regions which harbor most of the point mutations reside in the core domain. Large T antigen (Tag) of SV40 binds to p53 at the core domain and this binding results in the inactivation of p53 protein. Crystalline structure of three core domain molecules bound to p53 responsive DNA element revealed a unique motif for DNA-protein interactions (Cho et
al, 1994) . A Zn* atom coordination is required for the proper contacts that
permit binding (Muller-Tiemann et al., 1998). Additionally, mutational hotspots in this domain are shown either to prevent necessary DNA-protein contacts or disrupt the overall 3-D structure of the domain. DNA sequences of target genes which are recognized by the core domain have a common motif; PuPuPuCA/TT/AGPyPyPy . This motif is found as inverted pairs 10-13 bp apart, and there can be multiple copies of this pair (Zambetti et al., 1992 ).
- Oligomerization (tetramerization) domain is defined to be between residues 320 to 360.This domain consist of one beta-sheet and one alpha-helix in an antiparallel V-shape. Dimerization occurs through beta-sheets, and these dimers dimerize with their interaction with alpha-helices. Thus, p53 tetramer
is a dimer of dimers (Clore et a l, 1994). Within the tetramerization domain, an amino acid sequence motif that codes for a nuclear exclusion signal (NES) is identified. This motif is shown to be enough for the transport of p53 molecules out of the nucleus, however it is not exposed (so, not functional) when p53 exist as tetramers. This suggests that tetramerization not only provides sequence specific DNA binding, but also nuclear retention (Stommel
e ta l, 1999).
- A non-specific nucleic acid binding domain (C-terminal domain) is localized between residues 330 to 393. This domain binds single-stranded (ss) DNA more efficiently than double-stranded DNA which is thought to be important in DNA damage recognition. The domain has an open structure resulting in protease sensitivity. Nine basic amino acids give the domain an overall basic character. An interesting property of this carboxyl-terminal domain is that it stericly or allostericly blocks the sequence-specific DNA binding activity of the core domain. This inhibition is abolished in vitro by phosphorylation of certain residues by kinases (CK II or DNA-PK) or by the binding of certain specific antibodies (Hupp et a l, 1994 ). Furthermore, three
R175
#
R248 D B IV V· ____ , ^ < i > ^ m---·acidic hydrophobic basic
CTD
Figure 1.4: Domain structure of p53, togather with mutational hotspots. (Gray
Boxes I-V: regions concerved among species; TA\ N-terminal
transantivation domain; PR\ proline-rich region; DB\ DNA-binding core domain; O: oligomezization domain; CTD\ C-terminal non-specific DNA-binding domain; n: nuclear localization signals; red arrows: mutational hotspots)
nuclear localization signals (NLS) are present near the C-terminal domain. One of these signals is conserved among species and more effective than the other two (Shaulsky et ah, 1990 ). Alternatively spliced p53 transcripts which give rise to shortened and different C-terminal domain proteins are also observed. These alternatively spliced variants are shown to bind DNA targets more effectively than their regular counterparts, probably due to the lack of inhibition normally imposed by the C-terminal domain (Kulesz ei al., 1994 ).
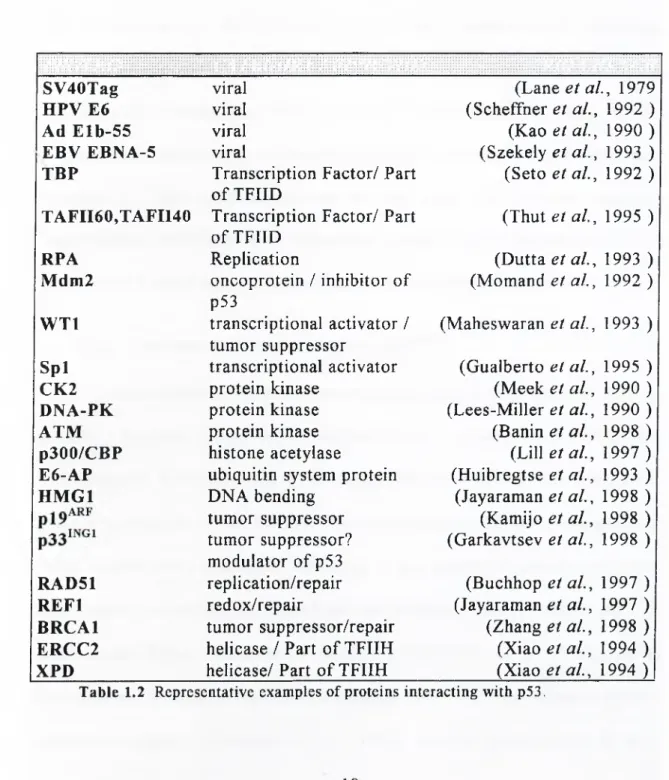
SV40Tag viral (Lane et al., 1979
HPV E6 viral (Scheffner et al., 1992 )
Ad Elb-55 viral (Kao e ta l, 1990 )
EBV EBNA-5 viral (Szekely et al., 1993 )
TBP Transcription Factor/ Part of TFIID
(Seto et al., 1992 ) TAFII60,TAFII40 Transcription Factor/ Part
of TFIID
(Thut e ta l, 1995 )
RPA Replication (Dutta et al., 1993 )
Mdm2 oncoprotein / inhibitor of p53 (Momand et al., 1992 ) WTl transcriptional activator / tumor suppressor (Maheswaran et al., 1993 ) Spl transcriptional activator (Gualberto et al., 1995 )
CK2 protein kinase (Meek et al., 1990 )
DNA-PK protein kinase (Lees-Miller et al., 1990 )
ATM protein kinase (Banin et ah, 1998 )
p300/CBP histone acetylase (Lill etal., 1997 ) E6-AP ubiquitin system protein (Huibregtse et ah, 1993 )
HMGl DNA bending (Jayaraman et ah, 1998 )
tumor suppressor (Kamijo et ah, 1998 ) tumor suppressor?
modulator of p53
(Garkavtsev et ah, 1998 ) RAD51 replication/repair (Buchhop et ah, 1997 )
REFl redox/repair (Jayaraman et ah, 1997 )
BRCAl tumor suppressor/repair (Zhang et ah, 1998 ) ERCC2 helicase / Part of TFIIH {Xxaoetah, 1994 )
XPD helicase/ Part of TFIIH {Xm oetah, 1994 )|
1.2.8 Interaction of p53 Protein with Other Proteins
p53 protein interacts with a variety of cellular and viral proteins. Although all of the domains contain binding sites for some proteins, most of the interactions are observed at either C- or N-terminal domains. These interactions are of utmost significance for both the execution of growth inhibitory roles of p53 and for the regulation of p53 itself , as discussed above. More than 10 viral and 40 cellular proteins are shown to interact with p53 in vitro and/or in vivo (see http:// w w w .dundee.ac.uk/ pathology /p53inter.htm, prepared by Peter A. Hall). Table 1.2 summarizes some of the representative interactions. It should be noted that, in some cases, physiological significance of these interactions remain to be established and presence of these interactions may be cell type- and situation specific. Nevertheless, identification of interaction partners of p53 has been, and will probably be a major step in the elucidation of p53-dependent pathways.
1.3 INGl Gene and its Protein Product, p33^”^*
A novel candidate tumor suppressor gene named INGl (for inhihiior o f
growth 1) has been cloned with a method based on substractive hybridization
(see Appendix A for more info.) This gene codes for a cDNA of 882 bp and a protein product of 33350 daltons. The nucleotide and protein sequence of INGl do not reveal significant homology to any known sequences and show only limited homology to retinoblastoma binding protein RbBP2 and to several zinc finger transcription factors (Garkavtsev et a l, 1996). The chromosomal location of the INGl is mapped to be 13q34 by radiation hybrid mapping technique (Zeremski et al., 1997) and by flourescence in situ
hybridization (Garkavtsev ei a l, 1997b). This sub-telomeric region of chromosome 13 is reported to be frequently rearranged in squamous carcinomas of head and neck.
Several line of evidence suggest that , the protein product of INGl, may be a tumor suppressor protein. Preliminary studies inferred that p33™°* may negatively regulate growth. Acute expression of transfected constructs encoding this gene inhibited growth in cell lines with a cell cycle arrest at Go/Gi phase while chronic expression of p33^’^‘^’' antisense RNA constructs promoted cell transformation. Additionally, inhibition of p33™^’ expression increases the frequency of focus formation in NIH 3T3 cells (Garkavtsev et al., 1996). Using indirect immunoflourescence, p33^’^‘^* protein was shown to be localized to nucleus, which is consistent with its role as a growth regulator (Garkavtsev et al., 1997b). Further studies reinforced the idea that p33‘*^^’ may be involved in cell cycle control like many other tumor suppressor genes. First of all, p33™°* is shown to be expressed at higher levels in senescent cultured human fibroblasts than in young, proliferation competent fibroblasts. Inhibition of p33™°' mRNA translation by introducing antisense constructs resulted in the extension of replicative life span of fibroblasts for several doublings (Garkavtsev et al., 1997a). Another property of p33^’^°^ is its role in the regulation of apoptosis. Serum deprivation of cultured cells leads to apoptosis at a much higher rate in cells infected with sense constructs of p33^^°’ than control cells. Conversely, apoptosis is prevented to significant extent when cells are transfected with antisense construct of p33’^^'’ (Helbing et al., 1997). These results are in parallel to
modes of actions of some tumor suppressor genes, such as p53, which can exert their antioncogenic functions by positively regulating apoptotic pathways. Another similarity observed between p33™°‘ and p53 is that growth inhibitory functions of both proteins are blocked by binding of SV40 large T- antigen (Garkavtsev et al., 1997a). Limited analysis of cell lines revealed a truncated protein in one of the neuroblastoma cell lines examined. The breast cancer cell lines examined showed a reduced expression of p33^'^‘^' compared to normal breast epithelial cell lines. The data accumulated until now suggests that p33™°‘ has a potential to act as a tumor suppressor protein.
A homologue of p33^^°* has been identified by a search of a database of randomly sequenced human cDNAs. The new gene is cloned and named INGIL C'INGl-like’’) (see Appendix B for details) . INGIL gene is mapped to 4q35.1 by FISH and radiation hybrid mapping techniques. INGIL cDNA shows 60.0% homology to that of INGl and the conceptual translation gives a polypeptide of 280 amino acids with 58,9% homology to p33‘'^^* (see Appendix C). Northern blot analyses revealed that INGIL is ubiquitously expressed in various human tissues and there is a relative increase in the mRNA levels of INGIL in colon cancer tissues compared to normal counterparts (Shimada et al., 1998 ).
C-terminal regions of INGIL and p33*^^* show very high homology and this region presents a zinc-finger motif of PHD (Plant HomeoDomain) type (see Appendix C). PHD type zinc-finger proteins are reported to be involved in transcriptional regulation, especially chromatin mediated transcriptional regulation (for review, see Aasland, 1995).
Another study reports that cooperates with p53 in cell growth control (Garkavtsev et ah, 1998 ). The similarities between p33^'^‘^* and p53, as mentioned above inspired Garkavtsev and colleagues to investigate a possible link between the functions of these proteins. First of all, a physical interaction between these two proteins was shown with coimmunoprecipitation. Additionally, it was proposed that growth inhibitory functions of these proteins are interdependent: none of them can function in the absence of the other. Furthermore, activation of transcription from the p2iWAFi pfojj^Qtej-^ a j^gy mechanism of p53-mediated growth control, depends on the expression of p33^^®* (Garkavtsev et al., 1998 ).Above results place P33ING1 signaling pathway for regulation of cell growth.
1.4 Aim and Scope of the Project
This study aims at identification of the cellular functions of p33™*^' protein through p53. For this purpose, mapping of the interacting regions of p53 with p33™°* protein has been investigated, using an in vitro method, namely GST pulldown assay. In conjunction with the mapping study, polyclonal rabbit antibodies against p33*^®’ has been raised for further studies, such as detection of expression levels of the p33*^‘^'' protein.
Such a study is thought to highlight the significant points in the interaction between the mentioned proteins. Comparison of binding regions in p53 with the regions that bind previously identified proteins may give clues to the functional importance of this interaction. Furthermore, interaction might have been mapped to a region where p53 mutations are prevalent, giving
CHAPTER 2: MATERIALS AND METHODS
2.1 MATERIALS
2.1.1 Chemicals
The laboratory chemicals were analytical grade and supplied from Sigma Biosciences Chemical Company Ltd. (St. Louis, MO, U.S.A) or from Carlo-Erba (Milano, Italy).
2.1.2 Bacterial Strains
The bacterial strain used in this work had the following genotypes:
E. colt, DH5a:
F', (f80dE(/flcZ)M15), recAl, endk\, gyrA96, (hi},hsdR\7,(T-
km-k), supE44,relAl, deoR, E(lacZYA-ar gF)U169
2.1.3 Enzymes
Restriction endonucleases were purchased either from MBI FERMENT AS Inc. (NY, U.S.A) ( Sail, Xbal, Xho , BamHl) or from Appligene-Oncor (Illkirch, France) (Hindlll, Sad).
Cloned Pfu Polymesase was purchased from Stratagene (CA, U.S.A). T4 Ligase was purchased from MBI FERMENT AS Inc. (NY, U.S.A). RibonuclaseA, thrombin and lysozyme were purchased from Sigma Biosciences Chemical Company Ltd. (St. Louis, MO, U.S.A).
The oligonucleotide primers used for the PCR amplification of p33‘^"^' were synthesized in the Beckman Oligo lOOOM DNA synthesizer (Beckman Instruments Inc. CA. U.S.A) at the Bilkent University, Faculty of Science, Department of Molecular Biology and Genetics, (Ankara, Turkey).
The primers are:
Forward: 5’- AGACGICGACAAATGTTGAGTCCTGCCAACG -3’
Sail site
Reverse: 5’- AGACAAGCTTCTACCTGTTGTAAGCCTCTC -3’
Hindlll site
Prior to synthesis, the primers were optimized for their annealing temperature, %GC content, hairpin and dimer formation potential with the help of Primer Designer Version2.0 computer program (Scientific & Educational Software, 1990,1991). It was confirmed by the Clone Manager Version 4.0 computer program (Scientific & Educational Software) that p3^iNGi contained no apparent Sail and Hindlll restriction enzyme recognition sequences.
2.1.5 Cloning Vectors
pGEX 2TK-P is a modified version of pGEX 2TK expression vector (GenBank #U 13851) of Pharmacia Biotech. (Uppsala, Sweden) kindly provided by Dr. U.Yavuzer. Proteins overexpressed from pGEX vectors exist as fusion proteins with GST (Glutathione S-transferase) enzyme which has a high affinity for glutathione. This property is exploited in the affinity purification of GST-fusion proteins using glutathione-linked beads (i.e., agarose or sepharose).
pBluescript II KS" (GenBank #X52326) is a phagemid that permits
expression of cloned genes in bacterial systems or in vitro transcription- translation (IVTT) reactions in eukaryotic systems. The vector contains T3 and T7 viral promoters that span the multiple cloning site and are oppositely oriented. These promoters enable eukaroytic expression of the cloned gene in the presence of respective viral RNA poymerases.
Navi\ (3-i
Figure 2.1 Vector map of pBluescript II
KS-2.1.6 Antibodies
The primary antibodies used in Western Blotting are as follows: Polyclonal Rabbit-anti-GSTp33 antibody (our production), monoclonal rabbit- anti-GST antibody (a gift from Dr. J.B. Crechet), monoclonal mouse-anti-p53 antibodies ( 7D3; specific to amino acids 211-220 ; HR221: specific to amino acids 371-380) kindly provided by Dr. Esma Yolcu.
Secondary antibodies used in Western Blotting are either Goat-anti- Rabbit antibodies or Rabbit-anti-Mouse antibodies both of which are Horse
Reddish Peroxidase (HPR) conjugated to permit visualization by chemilluminance.
2.1.7 Commercially Available Kits
QIAquick PCR Purification Kit (by Qiagen (Chatsworth, CA,
U S A)) is used for cleaning nucleic acids from contaminants.
QIAEXII (by Qiagen (Chatsworth, CA, U.S.A)) is for extraction of
DNA bands from agarose gels.
QIAGEN Plasmid Purification Kit (by Qiagen (Chatsworth, CA,
U.S.A)) is utilized to obtain plasmid DNA at higher amounts and concentrations with higher purity than those achieved by MiniPrep.
TNT Coupled Reticulocyte Lysate System is purchased from
Promega (Madison, WI, USA) for the single step in vitro transcription- translation (IVTT) of the cloned genes in an eukaryotic system.
Enhanced Chemiluminicance (ECL) System from Amersham
(Uppsala, Sweden) provides the means to visualize the HRP conjugated antibodies in Western Blotting.
Amplify from Amersham (Uppsala, Sweden) is a flourimetric method
to enhance the visualization of the signals generated by radioisotopes on polyacrylamide gels.
2.1.8 Apparatus
Vertical mini gel apparatus for polyacrylamide gel electrophoresis and the power supply are products of E-C Apparatus Corp. (Florida, U.S.A).
Horizontal midi gel apparatus used for agarose gel electrophoresis is purchased from Stratagene. Thermal cycler for PCR is a product of Perkin Elmer (CA, USA). Slab Gel Dryer is from Savant Instruments Inc.(N.Y.,USA). Semi-dry transfer unit for Western Blotting and GelDoc2000 Image analyzer for agarose gels are purchased from Bio Rad Laboratories (CA, U.S.A).
2.1.9 Materials for Protein Purification
Glutathione-Sepharose and Glutathione-Agarose resins
(Pharmacia Biotech (Uppsala,Sweden) enable the purification of GTS-fusion proteins and GST Pull-down assays by the high affinity of GST for its substrate, glutathione.
Aquaside (from Calbiochem, CA,USA) and the dialysis bags are kindly provided by Dr. A. Parmeggiani.
Proteases inhibitors used are either Protease Inhibitor Cocktail tablets or PefaBlock from Bbhringer-Mannheim, both of which are kindly provided by Dr.J.B. Crechet.
2.1.10 Materials for Autoradiograghy
Radiolabelled methionine (^^S-Met.) and light-proof film cassette (Hypercassette) and film developing unit are products of Amersham (Uppsala, Sweden). Medical x-ray films are purchased from Fuji (Tokyo, Japan).
As a DNA size marker, 1 kb DNA Ladder from MBI FERMENTAS Inc. (NY, U.S.A)is used.
SDS-PAGE protein size markers are either from Pharmacia (Uppsala, Sweden) or Bio Rad Laboratories (CA, U.S.A) (prestained markers). Migration patterns of these markers are given below;
2.1.11 DNA and Protein Size Markers
206 U5 79.5 49.5 Myosin â i ù « l ^ a ^ i o s W a s e
T*!iF Bovine serum sibumin Ovalbumin
34.8 Carbonic anhydrase
28.3 Soybean trypsin inhibitor
20.4 7.2 Aprolinin 94 67 43 30 20 ( a ) ( b ) ( c )
Figure 2.2 M igration patterns of size markers: a. Ikb DNA ladder (sizes in bp); b.
Prestained protein size marker, broad range; c. Protein size marker, low
molecular weight (sizes in kD)
2.1.12 Tissue Culture Reagents and Apparatus
Dulbecco’s modified Eagle’s medium (DMEM) and Trypsin/EDTA was obtained from Sigma Biosciences Chemical Company Ltd. (St. Louis, MO, U S A). FCS, Penicillin / Streptomycin mixture, Non-essential amino acids were obtained from Seromed. L-glutamine was from Biological Industries (Haemel, Israel). Tissue culture flasks, petri dishes, 15 ml polycarbonate centrifuge tubes with lids and cryotubes were purchased from Costar Corp. (Cambridge, England)
2.2 SOLUTIONS AND MEDIA
2.2.1 Agarose Gel Electrophoresis Solutions
Tris-acetic acid-EDTA (TAE) 40mM Tris-acetate ImM EDTA
Ethidium bromide: 10 mg/ml in water (stock solution), 30 ng/ml (working solution)
lx Gel loading buffer: 0.25% bromophenol blue, 0.25% xylene cyanol, 50% glycerol, ImM EDTA
2.2.2 Solutions for Plasmid DNA Isolation (MiniPrep)
Solution I 50 mM Glucose, 25 mM Tris.Cl, pH 8.0, lOM EDTA. Sterilize in autoclave.
Solution II Solution III
0.2 N NaOH, 1% (wt/vol) SDS 3 M Potassium acetate, pH 4.8 11.5% (v/v) glacial acetic acid
Phenol/ Chloroform 100%equilibrated phenol / 100% chloroform
2.2.3 Solutions for Bacterial Transformation
CaCb 50 mM in double distilled water,
filter sterilized
2.2.4 Microbiological media and antibiotics
Luria-Bertani medium (LB) Per liter: 10 g nutrient broth, 5 g bacto- yeast extract, 8 g NaCl, 0.52g Tris Base
For LB-agar plates, add 15 g/L bacto agar.
Sterilized by autoclave.
Ampicillin working solution; 100 |ig/ml or 50 pg/ml stock solution :100 mg/ml in ddH20
(stored at -20°C)
Kanamycin working solution: 25 |ug/ml
stock solution: 25 mg/ml solution in ddH20
(stored at -20°C)
2.2.5 Polyacrylamide Gel Electrophoresis Solutions
Resolving (Lower) Buffer (2X) 375mM TrisHCl, 0.2% SDS, pH 8.9
Stacking (Upper) Buffer (2X): 250mM TrisHCl, 0.2% SDS, pH6.8
Running Buffer (1X) 25mM Tris base, 192 mM glycine, 1% SDS
Sample Dye (2X) 50mM TrisHCl pH6.8, 1% SDS,2mM EDTA, 1% 2-ME, 0.02% Bromophenol Blue, 10% glycerol
Acrylamide-Bisacrylamide mix 24% acrylamide, 0.64% bisacrylamide
Amoniumpersulphate 10% (w/v) amoniumpersulphate in ddH20
Coomassie Staining Solution Solution 1: 10% Acetic Acid
Mix Solution 1 and 2 at a ratio o f 1:1
Destain Solution 14% ethanol, 7% acetic acid in ddH20
2.2.6 Protein Purification Buffers
Sonication Buffer 50mM Na-Phosphate Buffer (90% dibasic,
10% monobasic), 3OOmM NaCl, 10% (v/v) Glycerol, pH 7.8
Dialysis Buffer 20mM TrisHCl (pH 7.8), lOOmM NH4C1, 50% (v/v) glycerol, 5mM 2-ME
2.2.7 Solutions for Protein Concentration Estimation
Bradford Stock Solution: 100 ml of 95% ethanol, 200 ml of 88% phosphoric acid, 350 mg Serva Blue G. Filtered and stored in dark.
Bradford Working Solution; 21.25 ml of distilled water, 0.75 ml of 95% ethanol, 1.5 ml of 88% phosphoric acid, 1.5 ml of Bradford stock solution. Filtered.
Protein standard: 1.34 mg/ml bovine serum albumin (BSA) in double-distilled water. Stored at -20°C
2.2.8 Western Blotting Solutions
Transfer Buffer (pH 9.2)
20% (v/v)
48mM Tris Base, 39mM Glycine, Methanol 0.075% (w/v) SDS.
pH should be 9.2, but not adjusted
Phosphate Buffered Saline (PBS)(1X)
150mM NaCl, 16mM Na2HP04, 4mM NaH2P04,pH 7.5
PBS-T 0.1% (v/v) Tween-20 in PBS
2.3 METHODS
PCRs were performed using Cloned Pfu DNA Polymerase from Stratagene (La Jolla, CA, U.S.A) as described in the the manual provided by the supplier with some modifications. Ingredients of each 50 pi reaction tube is as follows: Cloned Pfu DNA Polymerase (2.5 units), dNTP mix from MBI FERMENTAS Inc. (NY, U.S.A) (10 nmoles), buffer of Cloned Pfu DNA Polymerase (lx), specific forward and reverse primers (250 ng or 125 ng each), DNA template (MCF7 total cDNA), ddH20 (making the total volume 50pl). Care was taken not to contaminate the reaction tubes with foreign DNA. Every time, a negative control reaction that contains no DNA template was set up. High extension times (2 min / cycle) were applied to compensate the low nucleotide incorporation rate of the enzyme. Polymerase was added into the reaction tubes after the initial denaturing step to disallow premature extention (“Hot Start PCR”). PCR products are checked for length and purity by AGE.
2.3.1 PCR amplification of cDNA
2.3.2 Agarose gel electrophoresis of DNA
DNA fragments were fractionated by horizontal electrophoresis by using standard buffers and solutions. DNA fragments less than 1 kb were generally separated on 1.0 % agarose gel, those greater than 1 kb were separated on 0.8 % agarose gels.
buffer to required percentage in microwave and ethidium bromide was added to final concentration of 30 pg/ml. The DNA samples were mixed with one volume loading buffer and loaded onto gels. The gel was run at room temperature in lx TAE at different voltage and time depending on the size of the fragments.
Nucleic acids were visualized under ultraviolet light (long wave, 340 nm) and standard DNA size marker, 1 kb DNA ladder, was used to estimate the fragment sizes.
2.3.3 Recovery of DNA Fragments from Agarose Gels
DNA fragments were extracted from agarose gels by using the QIAEX II gel extraction kit according to the manufacturer’s instructions.
Gel purification with the QIAEX II kit yields 60-70 % recovery of DNA fragments between 1.0 kb to 6.0 kb in 10-20 pi volume.
2.3.4 Methods for Cloning and Subcloning of Genes 2.3.4.1 Restriction Enzyme Digestion of DNA
Restriction enzyme digestions were routinely performed in 20 pi reaction volume for 1.5 to 4 hours and typically 1-10 pg DNA and 5-15 units of restriction enzymes were used. Reactions were carried out with the appropriate reaction buffer and conditions according to manufacturer’s recommendations.
simultaneously was generally possible by using a universal buffer (potassium acetate buffer) that fulfills the activity requirements of both enzymes. In this regard, YVTango Buffer from MBI Fermestas was usually suitable. The working concentration of this buffer was modified for each enzyme (either lx or 2x), and this way, it was possible to match the buffer requirements of various enzymes simultaneously.
2.3.4.2 DNA Ligation Reactions
DNA fragments were ligated into plasmid vectors in 10 pi reaction volumes containing 0.3-1.0 pg of linearized plasmid vector and 3-5 times molar excess of insert DNA in the presence of 1-4 Weiss units of T4 ligase and lx concentration of the standard ligation buffer supplied by with the enzyme. The reaction mix was incubated at either room temperature for 2.5-4 hours or at 16"C overnight.
2.3.4.3 Transformation of E .co li
Transformation of plasmid DNA into E.coli was achieved by using calcium chloride method. The following procedure is based on Ausubel et al. (1991).
P rep a ra tio n o f co m p e te n t cells
500 pi of E.coli glycerol stock solution was inoculated into 5 ml of LB medium containing selective agent and cells were grown at 37°C, shaking at 200 rpm to an optical density at 590 nm of 0.4 (approximately for 3 h). 1.5 ml of growing cells were centrifuged at 13,000 rpm for 1 min and gently resuspended in 500 pi ice-cold 50 mM CaCb.
T ra n sfo rm a tio n
Competent cells that were suspended in 500 pi ice-cold 50 mM CaCb were centrifuged at 13,000 rpm for 1 min. The pellet was resuspended gently in 100 pi of ice-cold 50 mM CaCb. 0.5-1 pi plasmid ( or the entire lOpl ligation mixture) was mixed with the competent cells and incubated on ice for 30 min. The competent cells were heat-shocked at 42°C for 90 seconds and the cells were then incubated on ice for 2 min. 1 ml of LB medium was added onto competent cells and incubated at 37°C for 1 h to allow the expression of antibiotic resistance gene before plating. After the incubation, 100 pi of transformation mixture was plated onto LB agar plates containing 50 pg/ml ampicillin or 25 pg/ml kanamycin to provide a selection for positive colonies carrying the newly introduced antibiotic resistance gene via transformed plasmid and incubated at 37°C overnight for the selection of antibiotic resistant tranformants. On the following day, single colonies were picked up either with a loop or a plastic micropippette tip under sterile conditions and innoculated into 5 ml of LB which contains the appropriate selective agent.
2.3.4.4 Plasmid DNA Preparation
S m a ll sc a le iso la tio n o f p la s m id D N A (M iniP rep)
The transformant bacteria strain containing the plasmid of interest was grown in 5 ml LB medium containing 100 pg/ml ampicillin at 37°C, while shaking at 200 rpm overnight. 1.5 ml culture was pelleted in 1.5 ml microfuge at 13,000 rpm for 2 min. After removal of supernatant, the cell were