Hanein-1, a novel conserved eukaryotic protein ubiquitously expressed in human tissues
Tam metin
Şekil
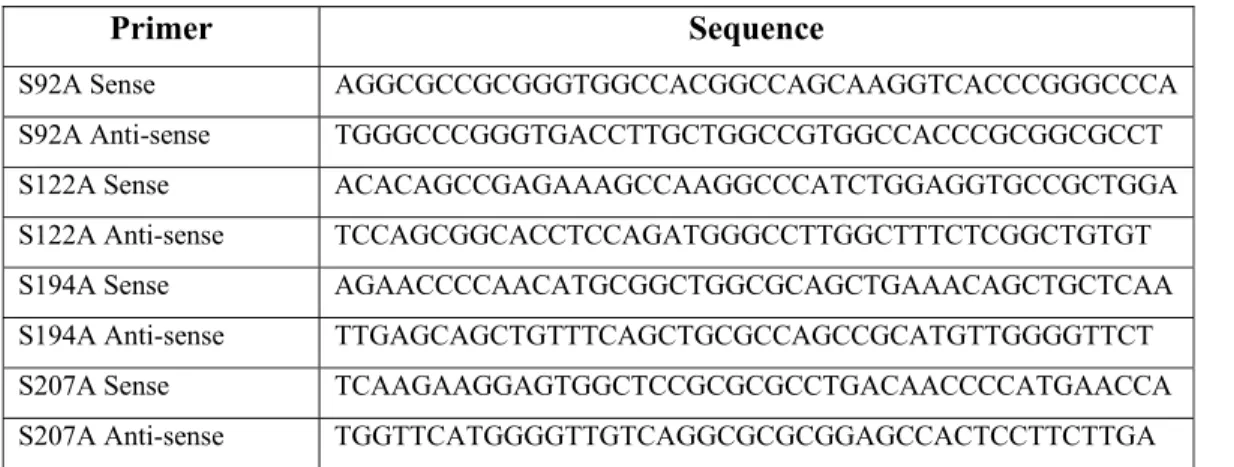





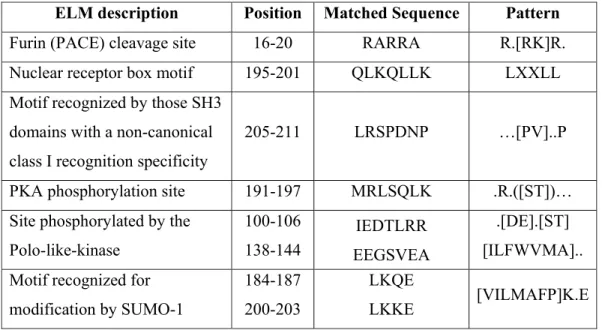
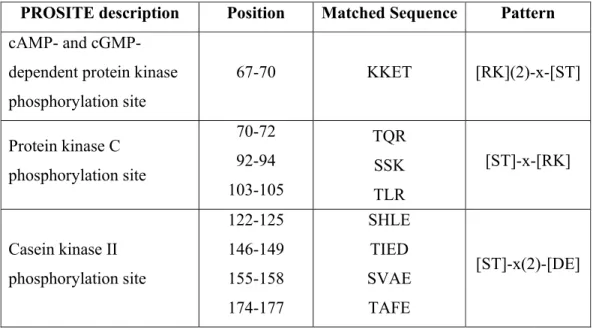

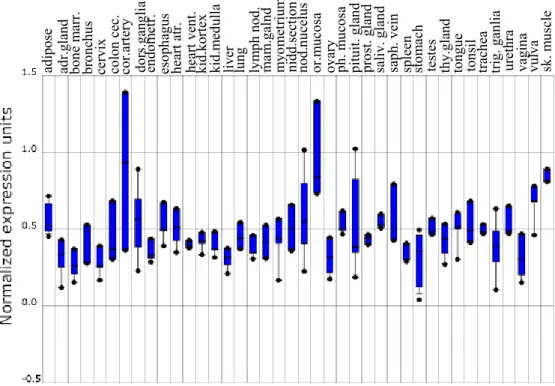
Benzer Belgeler
In our cases also, there was a history of cough, fever, neck pain and limited neck movement, and the examination revealed that subcutaneous emphysema swelling
Bu çalışmada karadut suyuna farklı sıcaklık ve sürelerde ısıl işlem uygulamasına bağlı olarak askorbik asit, tiamin, riboflavin, niasin ve toplam fenolik
附醫志工合唱團成立 10 週年暨幸安合唱團成立 17 週年之「聯合音樂會」 臺北醫學大學附設醫院志工合唱團,為慶祝成立 10 周年,特別結合創團
The fact that the dimension function of an algebraic n-homology sphere satisfies the Borel–Smith conditions suggests that more of the classical results on finite group actions on
Table 5.7: Number of transcripts coding for a single domain and those coding for multiple domains
A study of CaCo-2 human colon adenocarcinoma cell line reported that the 5-μM concentration of quercetin downregulates the expression of cell cycle genes,
In [11], for domains with a regular boundary we have constructed the approximants directly as the nth partial sums of p-Faber polynomial series of f ∈ E p (G), and later applying
In this Letter, given a number of nonthermal quantum channels, we study the problem of how to integrate them in a thermal engine so as to distill a maximum amount of work.. We
