T.C.
CLE ÜN VERS TES
TIP FAKÜLTES Ç HASTALIKLARI ANAB M DALI
CLE ÜN VERS TES HASTANELER NE BA VURAN
AT
K YERLE
ML VE/VEYA GENÇ TROMBOZ
TANILI HASTALARDA ETYOLOJ K FAKTÖRLER N
DE ERLEND
LMES
Dr. Ferhat B NGÖL UZMANLIK TEZ
TEZ DANI MANI
Prof. Dr. Mehmet Orhan AYYILDIZ
T.C.
CLE ÜN VERS TES
TIP FAKÜLTES Ç HASTALIKLARI ANAB M DALI
D CLE ÜN VERS TES HASTANELER NE BA VURAN
AT
K YERLE
ML VE/VEYA GENÇ TROMBOZ
TANILI HASTALARDA ETYOLOJ K FAKTÖRLER N
DE ERLEND
LMES
Dr. Ferhat B NGÖL UZMANLIK TEZ
TEZ DANI MANI
Prof. Dr. Mehmet Orhan AYYILDIZ
i ÖNSÖZ
Bilimsel dü ünme ve çal may bizlere ö reten, engin bilgi ve birikimlerini bizimle payla an, bugünlere gelmemizde büyük eme i olan, hekimli i bizlere
reten de erli hocam z Prof. Dr. Ekrem MÜFTÜO LU’na ba ta olmak üzere, ç Hastal klar A.B.D. Ba kan z Prof. Dr. M. Emin YILMAZ’a yeti memde büyük emekleri olan bütün de erli ö retim üyeleri; Prof. Dr. Orhan AYYILDIZ, Prof. Dr. Muhsin KAYA, Prof. Dr. Ali Kemal KAD RO LU, Prof. Dr. Kendal YALÇIN, Prof. Dr. Abdurrahman I IKDO AN, Prof. Dr. Alpaslan Kemal TUZCU, Doç. Dr. M. Ali KAPLAN, Doç Dr. Mehmet KÜÇÜKÖNER, Doç. Dr .Zülfikar YILMAZ, Yrd. Doç. Dr. Faruk KILINÇ, Yrd. Doç. Dr. Ya ar YILDIRIM, Yrd. Doç. Dr. Zuhat URAKÇI, Uz. Dr. Abdullah KARAKU , Uz. Dr. Elif Tu ba TUNCEL, Uz. Dr. Mazhar Müslüm TUNA, Uz. Dr. Zafer PEKKOLAY, Uz. Dr. Hikmet SOYLU, Uz. Dr. Ali Veysel KARA, Uz. Dr. Hüseyin KAÇMAZ, Uz. Dr. Zeynep ORUÇ ve rotasyon e itimim s ras nda bilgilerini benden esirgemeyen Kardiyoloji A.B.D, Enfeksiyon Hastal klar ve Mikrobiyoloji A.B.D, Gö üs Hastal klar A.B.D,
Radyoloji A.B.D ö retim üyelerine, birlikte çal maktan her zaman büyük mutluluk ve onur duydu um tüm asistan arkada lar ma ve ç Hastal klar A.B.D çal anlar na te ekkür ederim. Bugünlere gelmemde büyük eme i geçen aileme ve hayat arkada lar m e im Pelda ve o lum C wan’a te ekkürlerimi sunar m.
ii ÖZET
Atipik yerle imli trombozlar ve genç (40 ya ve alt ) trombozlarda etyolojik faktörlerin de erlendirilmesi
Giri ve amaç: Antikoagülan ve prokoagülan dengenin kal tsal veya edinsel bir faktör etkisiyle bozulmas ile ortaya ç kan tromboz, yerle im yerine göre semptomlar verir. Trombozlar, önemli morbidite ve mortalite nedenidir. Erken ya ta ve beklenen yerlerin d nda saptanan trombozlar atipik trombozlar olarak tan mlanmaktad r. Herediter trombofililer erken ya ta, anormal yerle imli veya s k tekrarlayan tromboz ataklar na neden olabilirler. Tromboz etyolojisinde APC direnci, protein-C ve protein-S eksiklikleri, AntitrombinIII eksikli i, PA -1 gen mutasyonu, faktör-V leiden mutasyonu ve protrombin gen mutasyonu gibi kal tsal faktörlerin yan nda travma, gebelik, immobilite, kanser, PNH, myeloproliferatif hastal klar, cerrahi, hipertansiyon, diyabet gibi edinsel risk faktörleri, tek ba na yada kal tsal risk faktörleri ile birlikte çoklu risk faktörü olarak bir arada bulunabilirler. Bu çal mada atipik yerle imli ve genç (40 ya ve alt ) tromboz tan hastalarda etyolojik risk faktörlerini belirlemeyi amaçlad k.
Materyal –metod: Çal maya 50 hasta (32K/18E) al nd . Hasta dosyalar taranarak retrospektif yap lan çal mada; 40 ya alt tüm trombozlar ve herhangi bir ya aral nda atipik lokalizasyonlu trombozu bulunan hastalar seçildi. Çal maya 16 ya alt hasta dahil edilmedi. Hastalarda atipik trombozlarda öne ç kan etyolojik risk faktörleri kal tsal ve edinsel nedenler aç ndan tarand . Tromboz lokalizasyonlar na göre öne ç kan risk faktörleri ara ld . Çal mada elde edilen sonuçlar n istatistiksel analizleri SPSS (statistical package for social scienses) for Windows 18. program kullan larak yap ld Verilerin de erlendirilmesinde de kenlerin frekans tablolar olu turuldu. Sonuçlar ortalama+-SD ve yüzde olarak (%) verildi. Tan gruplar na göre say sal de kenlerin ortalama olarak farkl olup olmad ANOVA testi ile test edildi. Kategorik de kenler için de Ki-kare testi kullan ld . P < 0.05 istatistiksel olarak anlaml kabul edildi.
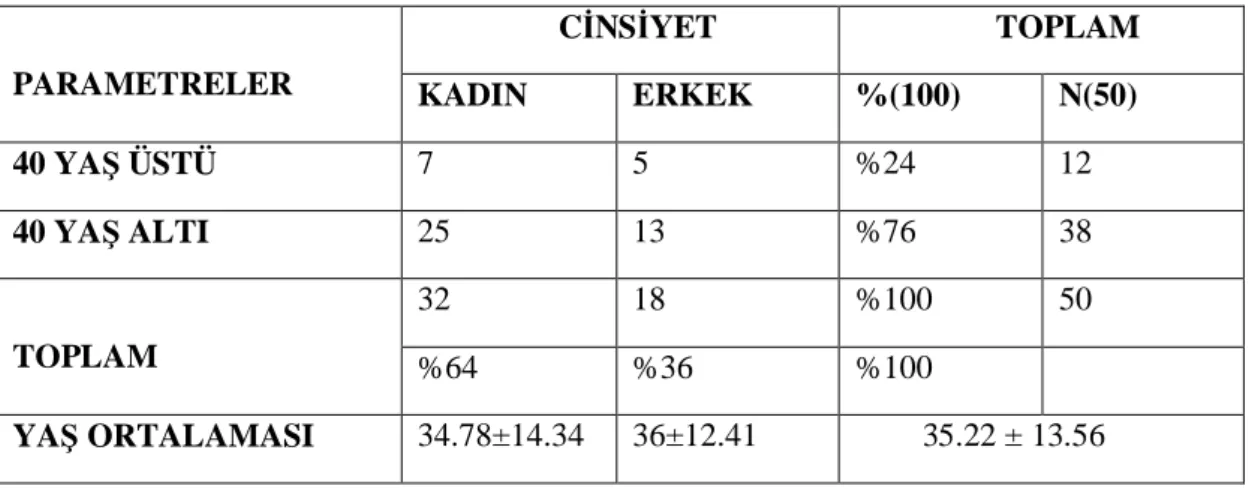
iii Bulgular: Çal maya; genç (40 ya alt ) tüm trombozlar ile herhangi bir ya aral nda atipik yerle imli toplam 32’si kad n (%64) ve 18’i erkek (%36)’ 50 hasta dahil edildi. Kad nlar n ya ortalamas ; 34.78±14.34. Erkeklerin ya ortalamas ; 36±12.41. tüm hastalar n ya ortalamas ; 35.22±13.56 olarak tespit edildi. Çal maya al nan hastalardan 12’si (%24) 40 ya üstü ve 38’s (%76) 40 ya alt saptand , 40 ya üstü hastalardan 5’i erkek (%10), 7’si kad n (%14), 40 ya alt hastalardan 13’ü erkek (%26), 25’i kad n (%50) olarak saptand . Çal maya al nan hastalar n 7’sinde (%14) OKS kullan sonras tromboz geli mi ti. Cerrahi sonras tromboz 6 (%12) hastada saptand , gebelik esnas nda 1 (%2) hastada tromboz geli mi ti, immobilzasyon sonras tromboz 1 (%2) hastada saptand , hipertansiyon 2 (%4) hastada saptand . MTHFR C677T 8 (%16) hastada homozigot ve 4 (%8) hastada heterozigot saptand , MTHFR A1298C 8 (%16) hastada homozigot ve 12 (%24) heterozigot ve 6 (%12) hastada birlikte heterozigot saptand . Hastalardan 7’sinde (%14) esansiyel trombositoz saptand ve 4 (%8) hastada JAK2 mutasyonu e lik ediyordu. Malignite öyküsü 1 (%2) hastada saptand , talasemi major 1 (%2) saptand , SLE tan 1 (%2) saptand , Fak-V leiden mutasyonu 3 (%6) hastada heterozigot saptand . Protein–S eksikli i 1 (%2) hastada saptand . AntitrombinIII eksikli i 2 (%4) hastada saptand . Protrombin gen mutasyonu 2 (%4) hastada heterozigot saptand , lupus antikoagulan 2 (%4) hastada yüksek saptand , fibrinojen 1 (%2) hastada yüksek saptand , PAI-1 (4G/5G) gen polimorfizmi 5 (%10) hastada saptand . Tan alan hastalarda kal tsal ve edinsel risk faktörleri 31 (%62) hastada bir arada saptand , hastalar n 6’s nda (%12) herhangi bir kal tsal ve edinsel risk faktörü saptanmad
Sonuç: Sonuç olarak atipik yerle imli ve genç ya ta görülen trombozlar; kal tsal ve edinsel etyolojik risk faktörlerinin bir arada saptanabildi i kompleks patolojik bir hastal k olup, abdominal bölge trombozlar n myeloproliferatif hastal klar aç ndan de erlendirilmesi ve kad nlarda OKS kullan n altta yatan kal tsal risk faktörlerinin varl nda tromboz riskini artt rd görüldü. Gebelik, lohusal k, OKS kullan gibi nedenlerden ötürü kad n cinsiyette daha fazla tromboz görüldü ü saptand .
iv ABSTRACT
Evaluation of etiological factors in young patient with atypically localized trombosis
Introduction: Thrombosis, which occurring with distortion on anticoagulant and procoagulant balance by a hereditary or acquired risk factor, causes symptoms according to the settlement place. Thrombosis is a major cause of morbidity and mortality. Thrombosis detected early and unexpected area is described as atypical thrombosis. Hereditary thrombophilia at an early age, can cause abnormal lacalized or recurrent attack of thrombosis. Inheritable factors such as protein-C and protein-S deficiencies, antithrombin III deficiency, PA -1 gene mutations, factor V Leiden mutation and mutation of prothrombin gene can be found in etiology of thrombosis and acquired factors such as trauma, pregnancy, immobility, cancer, PNH, myeloproliferative diseases, surgery, hypertension, diabetes can be found alone or with hereditary risk factors in etiology of thrombosis as multiple risk factors. Aim of this study is to determine the etiologic risk factors for atypical thrombosis.
Material-Method: 50 patients (32 F/ 18 M) were included in this study. Patient files were scanned in this study retrospectively; allless than 40 years of age with thrombosis and atypical localization of thrombosis in patients of any age range were chosen. Patients under 16 years of age excluded from the study. Etiologic risk factors for atypical thrombosis were screened for inherited and acquired causes. Prominent risk factors according to thrombosis localization were investigated. The statistical analyses of the results obtained from this study performed by using SPSS (statistical package for social science) for Windows 18. Variable frequency tables were created for evaluation of the data. Results were given as mean +-SD and percentage (%). According to diagnostic groups as to whether the different average of the numerical variables were tested by ANOVA. The Chi-Square test was used for categorical variables. P < 0.05 was considered statistically significant.
Findings: All thrombosis in young (under 40 year of age) and with all atypical localized thrombosis or recurrent in any age range of 32 women (64 %) and
v 18 men (36 %) patients were included this study. The mean of women’s ages; 34.78 ± 14.34. The mean of men’s ages; 36.22 ± 12.41. The mean of ages of all patients was 35.22±13.56. 12 of patients were over 40 years of age (24 %) and 38 of patients were under 40 years of age (76%) who included in this study. The 5 of over 40 years of age patients were male (10%) and 7 of them were female (14%). The 13 of under 40 years of age patients were male (26%) and 25 of them were female (50 %).
Patients enrolled in the study; MTHFR C677T 8 (16%) patients with homozygous and 4 (8%) were heterozygous patients, MTHFR A1298C 8 (16%) were homozygous and 12 patients (24%) heterozygous and 6 (12 %) were heterozygous with the patient. In 7 of the patients in this study (14%) it had developed thrombosis after oral contraceptive use. Thrombosis after surgery was found in 6 patients (12%). Thrombosis developed on 1 patient (2%) during pregnancy. Thrombosis after immobilization was detected in 1 patient (2%). Hypertension detected in 2 (4 %). Thrombocytosis was found in 7 patients (14%) and 4 (8%) of patients with JAK2 mutation was accompanied. Malignancy was seen in 1 patient (2%). Thalassemiamajor was seen in 1 (%2) patient. SLE diagnose was seen in 1 patient (2%). Fac-V Leiden Mutation was found in 3 patients (6%). Protein-S deficiency was seen in 1 patient (2%). Antithrombin III deficiency was seen on 2 patients (4%). Prothrombin gene mutation was found heterozygous in 2 patients (4%). Fibrinogen-I was found high in 1 patient (2%). Lupus Anticoagulant detected high in 2 patients (4%). PAI-1 gene mutation was detected in 5 (10%) patients. Hereditary and acquired risk factors were found together in 31 patients (62%). There was no hereditary and acquired risk factor in 6 of patients (12%).
Result: As a result, atypical thrombosis is a disease of complex etiology can detect a combination of hereditary and acquired risk factors. Abdominal thrombosis should be evaluated in term of myeloproliferative diseases. The using of oral contraceptives increase the risk of thrombosis in women with underlying genetical risk factors. It has detected thrombosis can seen in female more than male sex because of pregnancy, confinement or using oral contraceptives.
vi NDEK LER Sayfa No. ÖNSÖZ ... ÖZET ... ABSTRACT ... NDEK LER ... TABLOLAR L STES ... MGELER VE KISALTMALAR..……... 1. ve AMAÇ ... 2.GENEL B LG LER ... 2.1.Hemostaz ... 2.2.Tromboz ve Trombofili ... 2.2.1.Genel Bak ...
2.2.2. Kal tsal Trombofili Taramas Yap lmas Gereken Durumlar ... 2.2.3. Kal tsal Trombofili Taramas Yap lmas Önerilmeyen Durumlar ...
2.2.4. Kal tsal Trombofilinin Klinik Önemi ... 2.2.5. Edinsel Tromboz Nedenleri ...
2.2.6. Kal tsal Tromboz Nedenleri ... 2.2.6.1. Antitrombin III Eksikli i ... 2.2.6.2. Protein C Eksikli i ...
2.2.6.3. Protein S Eksikli i ... 2.2.6.4. Aktif Protein C (APC) Direnci ve Faktör V Leiden Mutasyon ..
2.2.6.5. Protrombin G20210A Mutasyonu ... 2.2.6.6. Hiperhomosisteinemi ... 2.2.6.7. Faktör VIII Yüksekli i ... 2.2.6.8. Fibrinojen Gen Mutasyonu ... 2.2.6.9. Plazminojen Aktivatör nhibitör 1(PAI–1) Gen Poliformizmi .. 2.2.6.10. Faktör XIII Gen Polimorfizmi ... 2.2.7. Atipik Trombozlar ... 2.2.7.1.Serebral Ve Sinüs Ven Trombozlar ...
2.2.7.2.Üst Extremite Ven Trombozlar ... 2.2.7.3.Juguler Ven Trombozu ...
2.2.7.4.Vena Cava Trombozu ... 2.2.7.5. nferior Vena Cava Trombozu ...
2.2.7.6.Portal Ven Trombozu ... 2.2.7.7.Hepatik Trombozu ... 2.2.7.8.Mesenterik Ven Trombozu ...
2.2.7.9.Splenik Ven Trombozu ...
2.2.7.10.Renal Ven Trombozu ... 2.2.7.11.Ovaryen Ven Trombozu ... 2.2.7.12.Penil Ven Trombozu ...
2.2.7.13. Alt Extremite Superfisial Ven Trombozu ... 2.2.7.14. Renal Ven Trombozlar ... 3. MATERYAL VE METOD ... 3.1.Hasta Seçimi ve D lama Kriterleri ... 3.2. Biyokimyasal Ölçümler ... 3.3. statistik ... i ii iv vi viii ix 1 3 3 5 5 8 8 9 9 10 13 14 15 16 18 18 19 20 21 22 23 23 24 25 25 25 26 27 27 27 28 28 29 29 29 31 31 31 32
vii 4. BULGULAR ... 5. TARTI MA ... 6. SONUÇ ... 7. KAYNAKLAR ... 33 42 47 48
viii TABLOLAR L STES
Sayfa No. Tablo 1: Kal tsal trombofili görülme s kl ve trombozla ili kisi ... Tablo 2: Nadir rastlanan primer trombofili nedenleri ... Tablo 3: Sekonder trombofili nedenleri ... Tablo 4: Protein C ve protein S eksikli i tipleri, ekilleri ve klinik tipleri ... Tablo 5: Çal maya al nan hastalar n demografik bulgular ... Tablo 6: Çal maya al nan hastalar n tan lara göre da ... Tablo 7: DVT tan hastalarda saptanan etyolojik risk faktörleri ... Tablo 8: Pulmoner tromboemboli tan hastalarda saptanan etyolojik risk
faktörleri ... Tablo 9: skemik inme tan hastalarda saptanan etyolojik risk faktörleri ... Tablo 10: Sinüs ven tromboz tan hastalarda saptanan etyolojik risk
faktörleri ... Tablo 11: Retinal ven trombozu ve iskemik inme tan hastalarda saptanan
etyolojik risk faktörleri ... Tablo 12: Hepatik ven tromboz tan hastalarda saptanan etyolojik risk
faktörleri ... Tablo 13: Portal ven tromboz tan hastalarda saptanan etyolojik risk faktörleri ... Tablo 14: Mesenterik ven tromboz tan hastalarda saptanan etyolojik risk faktörleri ... Tablo15: Splenik, V.C. , portal,-mesenterik ven tromboz tan hastalarda
saptanan etyolojik risk faktörleri ... Tablo 16: Juguler ven tromboz tan hastalarda saptanan etyolojik risk faktörleri ... Tablo 17: Retinal ven tromboz tan hastalarda saptanan etyolojik risk
faktörleri ...
Tablo18: Saptanan etyolojik risk faktörlerinin say sal da ... 11 12 12 15 33 34 35 35 36 36 37 37 38 38 39 39 40 41
ix MGELER VE KISALTMALAR
µm : Mikrometre
ADP : Adenozin Difosfat
APC : Aktive Protein C
APCR : Aktive Protein C Rezistans AT-III : Antitrombin III
ATP : Adenozin Trifosfat
Ca++ : Kalsiyum
DVT : Derin Ven Trombozu
EGF : Epidermal Growth Factor
GIa : Gama Karboksi Glutamik Asit
HMWK : Yüksek Molekül A rl kl Kininojen (High Moleküler Weight Kininojen, Fitzgerald Faktörü )
MTHFR : Metilentetrahidrofolat Redüktaz
ng : nanogram
NO : Azot Oksit
PAI–1 : Plazminojen Aktivatör nhibitörü–1
PC : Protein C
PIVKA : K vitamini yoklu unda olu an proteinler (Proteins Induced By Vitamin K Absence)
PK : Prekallikrein (Fletcher faktörü)
PS : Protein S
PTE : Pulmoner Tromboemboli
PTGM : Protrombin G20210A Mutasyonu
PVT : Portal Ven Trombozu
RES : Retikülo Endotelyal Sistem
TAT kompleksi : Trombin- Antitrombin III kompleksi
TF : Doku Faktörü (Tissue factor)
TFPI : Doku Faktör Yolu nhibitörü (Tissue Factor Pathway Inhibitör)
t-PA : Doku Plazminojen Aktivatörü
TXA2 : Tromboksan A2
VTE : Venöz Tromboemboli
1
1.G VE AMAÇ
Normal kan ak ; koagülan, antikoagülan ve fibrinolitik sistemler aras ndaki hassas dengenin korunmas ile sa lanmaktad r. Bu hemostatik denge, hem kanamay hem de uygun olmayan p ht olu umunu önlemektedir. Koagülasyon bozukluklar tromboembolik hastal klara yol açabilir.
Trombofili (Thrombo-philia: trombozu sevme) tromboza e ilim olu turan tablolar tan mlamakta kullan lan bir terimdir. Tromboz geli imi multifaktöriyeldir (1). Çok say da edinsel ve kal tsal faktör de ik mekanizmalarla tromboz olu umuna neden oldu u bilinmektedir (1-2). Arteriyel ve venöz sistemde tromboz formasyonunun farkl olmas , bu iki sistemde farkl etyolojilerin rol oynad dü ündürmektedir (arteryel platelet bak ndan, venöz ise fibrin bak ndan zengindir). Arteriyel sistemde endotel hasar ve trombositlerin fonksiyonel bozukluklar n önemli rol oynad , venöz sistemde ise daha çok staz ve p ht la ma sistemine ait bozukluklar n (p ht la ma sistemini kontrol eden do al inhibitör mekanizma bozukluklar n) tromboz geli imine neden oldu u bilinmektedir (3,4).
Venöz tromboz; venöz sistem içinde fizyolojik gereksinim olmadan p ht olu mas r. Ço unlukla alt extremitelerin derin venlerinde olu an trombuslar o ekstremitede lokal yak nmalara ve/veya bazen kopup pulmoner arter ve dallar
kayarak pulmoner tromboemboliye (PTE) neden olabilir. PTE’ si olan hastalar n %70’inde alt ekstremitede ço ulukla asemptomatik venöz trombus tespit edilmi tir (5).
Tromboz toplumda s k görülen hastal k olup, insan hayat nda önemli mortalite ve morbidite nedenlerinden biridir. Eri kinlerde insidans yakla k %1 iken, çocuklarda bu oran y lda 1/100.000 olarak belirtilmi tir (1,4). Herediter trombofili denilince akla tromboza e ilimi olan bireyler gelmektedir (2,6). Tromboz etyolojisi karma k olup genelde birden fazla faktör içerir. Genellikle herediter ve edinsel risk faktörlerinin etkile imi sonucu ortaya ç kar (1,2). Edinsel faktörler; cerrahi müdahale, obezite, cinsiyet, uzun süre hareketsiz kalmak, maligniteler, travma, nefrotik sendrom, myeloproliferatif hastal klar, PNH, konjestif kalp yetmezli i, östrojen kullan , hamilelik ve lohusal k gibi do rudan risk faktörleriyle ba lant ve ya la artan bir insidans gösterir (1, 7-9).
2 Yak n geçmi te tomboemboli ile ba vuran hastalar n bir k sm nda altta yatan nedenler bulunamamaktayd . 1993 y ndan önce kal tsal nedenler trombozlu olgular n %10-15’inde saptanabilmekteydi ve Protein C, Protein S, ATIII eksikli i ile s rl kalmaktayd (8,9). Son y llarda aktive protein C rezistans (1993), hiperhomosisteinemi (1994), faktör V Leiden mutasyonu (1994), protrombin G20210A mutasyonu (1995), plazminojen aktivatör inhibitörü–1 gen polimorfizmi, fibrinojen defektleri, faktör 13 gen mutasyonu, faktör 12 gen mutasyonu, plazminojen eksikli i, trombomodulin gen mutasyonlar , plazma proteinlerindeki anormalliklerin tespiti etyolojisi %40-60 ayd nlat lm r (11, 12). Kal tsal trombofili nedenlerini genetik olarak ta yan bireylerde tromboz riski artmakla birlikte ya am boyu hiç trombotik atak geçirmemeleride mümkündür. Veya bu hastalarda tekrarlayan trombotik ataklar aras nda uzun süre devam eden asemptomatik dönemler olabilmektedir. Bu durum, tek ba na kal tsal nedenlerin yeterli olmad , tromboz meydan gelmesinde baz edinsel faktörlerin katk oldu unuda göstermektedir (1, 10, 11, 13-14). Kal tsal trombofili tan için yap lacak testler oldukça zahmetlidir, pahal r ve uygun testler kullan lmazsa yan lt sonuçlar elde edilebilece inden titizlikle seçilmelidir (15). Çal mam zda amac z, atipik trombozlu hastalarda etyolojik risk faktörlerinin belirlemek, tromboz yerle im yerine göre öne ç kan etyolojik risk faktörlerini s flamak ve birden fazla risk faktörü olan hastalar tekrarlayan trombozlar aç ndan uyarmak ve uzun süre tedavi verilmesi gereken hastalar belirlemek.
3 2.GENEL B LG LER
Kan, organizmada kapal bir kanallar sistemi olan damarlar içinde dola an bir dokudur. Dola m kan , plazma ad verilen s bir ortam içinde süspansiyon halinde bulunan ekilli elemanlardan meydana gelmi tir. Ortalama bir ki inin kilogram bas na yakla k 70 ml (70ml/kg) kan vard r. Metabolizma, hormonlar yoluyla hücresel ileti im ve immün savunma, gaz transportu, dahil pek çok fizyolojik olayda kilit rol oynar (16, 17). Kan pH’ ortalama 7,4 ve dansitesi 1,035–1,075 g/cm
3
aras ndad r. Kan hacminin neredeyse %50-60’ s , geri kalan ise ekilli elemanlardan olu maktad r. Kan, ekilli elemanlar ve protein içeri i nedeniyle visközdür. Plazma ad verilen s bile enin yaklas k %90’ su; geri kalan %10’luk
sm ise aminoasitler, glukoz ve diger metabolitler, çe itli protein, iyonlar ve hormonlar taraf ndan olu turulur. Serum; plazman n, koagülasyon faktörleri ve fibrinojenin uzakla lmas ndan sonra geriye kalan k sm r (17, 18).
Kan, görev ald fizyolojik olaylar yerine getirebilmek için s halini sürdürmek zorundad r. Dola m sisteminde non koagüle ve ak kan olmas na katk da bulunan etkenler; antikoagülan, koagulan ve fibrinolitik sistemler aras ndaki hassas dengenin korunmas , damar sisteminin kas labilir olmas , kalbin pompalama gücü, gö üs bo lu undaki negatif bas nç ve venöz damarlarda kan n geri dönmesini engelleyen kapakç klar olarak say labilir. Kan damarlar ( arter, ven ve kapillerlerde ) histolojik katmanlar ve içerikleri aç ndan birbirlerinden farkl rlar. Damar kollajeni ba ca tip III ve tip I kollajen’den olu ur. Tip III kollajen trombositin yap mas en iyi ekilde sa lar ve damar n bütünlü ünü korur. Bunun eksikli inde (örne in Ehlers- Danlos sendromu tip IV) büyük bir arterin y rt lmas sonucunda ani ölüm s k görülür. Bunun d nda damar n iç yüzeyinde, endotel hücrelerinin olu turdu u kaygan yüzey de kan n ak kan ve s halde akmas nda önemli bir etkendir Elastin; damar n esnekli ini, bedenin hareketleri ve kalbin pompalama kuvvetiyle uyumu sa lar, düz kaslar uyar ya göre kas rlar (17, 19, 20).
2.1.Hemostaz
Hemostaz, kan n dola mda s halde kalmas sa lay p do ru ekilde akmas na neden olan mekanizmad r. Bu mekanizma kan damarlar nda bir travma
4 sonucu olu an kanamay durdurmas ve daha sonra ayn damar n fonksiyonunu devam ettirmesi için damar n p ht dan ar nd lmas durumlar da içerir (19, 21). Koagülasyon hemostaz n bir faz r. Yaralanmay takiben endotel, dola an trombositler ve koagülasyon faktörleri etkile imleri ile koagülasyon sa lan r.
Damar n hasar görmesi sonucu olu abilecek kanamay önleyen hemostaz sisteminin iki önemli özelli i vard r. Birincisi: yüzey ba ml yani hasarlanm bölgede aç a ç kan fosfolipidler ile hemostaz n ba lamas , olay n belli bir alanda rl kalmas sa lar. Trombosit yüzeyindeki fosfolipidler koagülasyon için birincil p ht la ma alan olu turur. kincisi ise çok say da negatif geri besleme yollar bulunmas r. Hemostazda görev alan her sistem, kar t görev gören bir inhibitör sistemle dengelenmi tir. Ço u kez bir sistem uyar ld nda kar t sistemini kendisi devreye sokar; bu sayede her sistem, kar t sistemi ile e zamanl harekete geçmi olur. Dolay yla kan n p ht la mas (hemostaz) ve p ht n erimesi (fibrinoliz) süreçleri birbiri ile yak ndan ili kilidir ve sürekli bir etkile im halindedir (17, 19).
Damarlara bir hasar meydana geldi inde üç olay ardarda meydana gelir (19); 1- Damarda, ba lang ç hasar n ard ndan büyük oranda refleks nörojen mekanizmalara ba olan ve endotelin (güçlü vazokonstriktör) gibi faktörlerin lokal sal ile geli en k sa süreli vazokonstriksiyon olur (vasküler faz).
2- Endotel zarar ayn anda, yüksek trombojenik etkiye sahip olan subendatelyal hücre d matriksi (ECM) de aç a ç kar r. Bu da trombositlerin yap mas na, aktive olmas na ve salg granüllerinin sal nmas na olanak sa lar. Sal nan bu maddeler daha fazla trombositleri bir araya getirerek (agregasyon) hemostatik t kac olu turur (Trombosit faz ). Bu da primer hemostaz i lemidir.
3- Hasar bölgesinden aç a ç kan doku faktörü, trombositlerden sal nan faktörlerle beraber etki ederek p ht la ma sistemini uyar r. Sonuçta fibrin birikimi olur. P ht la ma sisteminde aktive olan trombin de daha fazla trombositin toplanmas na ve granüllerin aç a ç kmas na yol açar (Plazma faz ). Sekonder hemostaz denilen bu dönem, trombosit t kac n olu mas ndan daha uzun sürer.
Özetle: damar hasar n oldu u bölgede trombositlerin t kaç olu turmas na primer hemostaz, bunu takiben koagülasyon sisteminin aktifle erek fibrin p ht olu turmas na sekonder hemostaz ad verilir.
5 2.2. Tromboz ve Trombofili
2.2.1. Genel Bak
Tromboz; vasküler sisteminde kan n ak kanl engelleyen, hemostatik sistemde antikoagülan-fibrinolitik sistem ile koagülasyon sistemi aras ndaki dengeninkal tsal veya edinsel nedenlerle bozulmas sonucu ortaya ç kan patolojik bir durumdur. Trombofili (Thrombo-philia: trombozu sevme) tromboza e ilim yaratan tablolar tan mlamakta kullan lan bir terimdir (2).
Vücudun bir savunma mekanizmas olarak tromboz geli ir. Vasküler bir zedelenme sonras fizyolojik artlarda antitrombotik olan endotelial bölgenin bütünlü ünün bozulmas ile subendotelial bölgeden aç a ç kan trombotik faktörlerin
kan kayb engellemek için; etkin ve çabuk bir cevab r. Meydana gelen tromboz t kaç görevini görür ve görevi sona erince fibrinolitik sistem taraf ndan eritilerek ortadan kald r.
Patologlar taraf ndan (Zahn, 1875; Bizzozero,1882; Eberth & Schimmelbusch, 1886) 19.yüzy lda trombusun temel yap aç k bir ekilde tan mlanm r. Bu patologlar trombusun; statik sistemde olu an kan p ht ndan tamamen farkl oldu unu ve aktif, sirküle halde olu tu unu ve bu olu umda fibrin a içinde trombositlerin ve polimorfnükleer lökositlerin seçici bir sekilde depoland göstermi lerdir (2, 7). Tromboz kan n ak s ras nda hümoral, selüler ve vasküler faktörlerin birbirleriyle etkile imi sonucu geli ir. Kinetik bir süreç olmas özelli iyle, statik bir fenomen olan kan p ht la mas ndan ay rt edilir (7).
Fizyopatolojik olarak Tromboz; çökelmeye neden olan etmene ba olmaks n, p ht la ma sisteminin tüm elemanlar içerir ve öyle özetlenebilir:
a) Trombositlerin tutunup y larak sal nd klar endotel ve subendotel bölgesinde, p ht la ma ba lar.
b) Olu an Fibrin p ht yla, fibrinolitik süreç boyunca normalde trombozu rland ran mekanizmalar aktive olur.
c) Patolojik trombus; antikoagulan, koagülan ve fibrinolitik süreçler aras ndaki dengesizli in sonucu olarak ortaya ç kar. Bu olu umdan sonra trombusun tamam ya da bir parças emboli yapar (24).
6 lk olarak 1856 y nda Alman patolog olan Rudolf Virchow, trombozun patogenezini aç klayan bir hipotez geli tirmi tir. Geli tirdi i bu hipoteze göre tromboz olu umunda üç ana faktör (Virchow’s triad ) rol oynamaktad r (2,7).
Bunlar; 1. Staz
2. Damar duvar nda hasar
3. Dola an kandaki anormalliklerdir.
Sonraki çal malarda tromboz geli iminde; kan ak , fibrin olu umu ve fibrin y aras ndaki hemostatik dengenin de önemli oldu u gösterilmi tir ve baz tromboza e ilim yapan etkenler tespit edilmi tir. Bunlar; damar duvar hasar , trombosit aktivasyonu, kan koagülasyon faktörlerinin aktivasyonu, kan ak nda yava lamas ve fibrinolizin inhibisyonudur. Tromboz olu umunu engelleyen sistemler ise serin proteazlar inhibe eden do al inhibitörlerin normal fonksiyon ve miktarda olmas , endotelin antikoagülan aktivitesi, retiküloendotelyal sistem ve hepatositler taraf ndan aktif proteazlar n temizlenmesi ve fibrinolitik sistemin sa lam çal mas r (2, 25, 26).
Trombofili (Hiperkoagülabilite); çe itli predispozan faktörlere veya antikoagülan yoldaki kal tsal defektlere ba olarak artm tromboz riskini tan mlar. Dola mda, p ht la ma inhibitörlerinin eksikli i (ATIII, PC, PS), p ht la ma faktörlerinin konsantrasyonlar n artmas (FVII, FVIII, FIX) ya da aktif faktörlerin bulunmas (aktif FIX) fibrinolitik aktivitede azalma, trombosit reaktivitesinde art ve trombositoz gibi degi iklikler hiperkoagülabilite örnekleri olarak say labilir (10, 14, 25). Tromboz edinsel faktörlere ba ysa sekonder, kal tsal predispozan faktörlere ba ysa primer hiperkoagülabilite olarak isimlendirilir. venöz tromboz geli mesinde kal tsal faktörlerin rolü daha fazlayken, arteriyel tromboz geli mesinde etkisi daha azd r. Arteriyel tromboz diffüz veya lokal atherosklerozun bir komplikasyonu olarak geli mektedir. Arteryel trombozlu hastalarda atheroskleroz olu umuna neden olan hipertansiyon, hiperlipidemi, diyabet, hiperhomosisteninemi, trombosit fonksiyonlar nda bozuklu a neden olan genetik bozukluklar önemlidir. Hiperkoagulabilite özellikle venöz trombozla ili kilidir.
Multifaktöriyel bir hastal k olan tromboz toplumda s k görülen bir hastal kt r. çocuklarda oran y lda 1/100000 iken, eri kinlerde insidans yaklas k % 1 olarak
7 saptanm r (1, 27). Etiyolojisi karma k olan tromboz genelde birden fazla faktör içerir. Ço u kez çevresel ve kal tsal risk faktörlerinin etkile imi sonucu ortaya ç kar (3, 6).
Bir arteriyel p ht incelendi inde trombositten zengin bir yap tespit edilir. Arteriyel trombozun en bilinen nedeni, aterosklerotik damar hastal takiben endotelde olu an zedelenmedir. Plak rüptürü geli ince trombositler damar yaralanmas olmu gibi adezyon, sekresyon ve agregasyon yaparak damar duvar ndaki defekti kapatmaya çal makta, ancak normal hemodinami ve endotelin trombosit yan dengeleyici rolü bozuldu u için trombosit yan abart olmaktad r. Sonuçta fibrin birikimi üzerine trombosit t kac olu maktad r. Genelde iliak arterler, dorsal aorta ve koroner arterler gibi geni ve orta ölçüdeki arterleri kapsar (28). Kaoagülasyon sisteminin daha fazla aktivasyonu, ilave fibrin birikimini ilerleterek damar n total t kanmas na neden olur. Arteriyel trombuslar; Beyaz trombus olarak bilinirler, fibrin ve trombosit tabakalar ndan olu mu olup, hasarl arter duvar na s bir ekilde ba lan rlar. Arteriyel trombozun en bilinen ekilleri, akut miyokard infarktüsü ve serebral vasküler olaylard r (10, 14, 29, 30).
Venöz tromboz, venöz sistem içinde normalin d nda p ht olu mas r. En önemli nedenleri; p ht la ma sistemine ait bozukluklar ve kan ak nda yava lama (staz) d r. Venöz trombus; genellikle baca n derin venlerinde, daha nadir olarak beyin, mezenter, retina ve karaci er venlerinde olu ur. Meydana gelen p ht lar bazen lokal semptomlara neden olsada bazen dola ma kat larak pulmoner arter yada dallar t kayarak ölümcül bir tablo olan pulmoner tromboemboliye neden olurlar.
ht yap nda; fibrin, eritrosit ve trombositlerin mevcudiyetinden bu yap ya k rm trombus ad da verilir (8, 10, 26, 31). Yap lan klinik çal smalarda ve toplum taramalar nda; venöz tromboembolizmin 100000/117 y l ki iyi etkileyen yayg n bir hastal k oldu u ve ya ile birlikte insidans n giderek artt gösterilmi tir. Ayr ca kal tsal tromboz nedenlerinin tamam na yak n venöz tromboza e ilim olusturdu unu göstermektedir. Genetik risk faktörleri ta yan bireylerde tromboz riski artmakla birlikte, herhangi bir trombotik atak geçirmemeleri de mümkündür. Bu durum kal tsal nedenlerin tek ba na yeterli olmad , kal tsal nedenlerin yan nda baz edinsel faktörlerin de katk oldu unu göstermektedir (14, 26, 28, 32).
8 2.2.2. Kal tsal Trombofili Taramas Yap lmas Gereken Durumlar
1. Ailede tromboz e ilimi olanlar (Ailede hastan n kendisi d nda 2 semptomatik VTE geçirmi birinci derece akraba daha bulunmas ve/veya hastan n ailesinde tekrarlayan idyopatik VTE hikâyesi bulunmas ).
2. lk venöz tromboemboli (VTE) ata 40 ya alt nda geçirenler
3. Serebral ve kar n içi ven t kan kl klar nda kal tsal trombofili taramas yap labilir; ancak kar n içi ven trombozlar nda öncelikle edinsel nedenler
lanmal r.
4. Purpura fulminans varl nda özellikle PC ve PS eksikli i taranmal r. 5. Venöz tromboz riski ta yan gebe kad nlar
6. Tekrarlayan idyopatik/minör tetikleyici etkene ba VTE öyküsü olan hastalar
7. Tromboza e ilimli ailelerde ve trombofili saptanmas n ki ide medikal yakla m de ikli ine yol açaca durumlarda aile taramas yap labilir.
8. Tromboza yatk n ailelerde yüksek riskli trombofilik bozukluklar taranabilir (33, 34).
2.2.3. Kal tsal Trombofili Taramas Yap lmas Önerilmeyen Durumlar 1. Heparine ba trombositopenide,
2. Majör geçici risk faktörleri, aktif kanser ve tromboza yol açabilen di er klinik durumlar (SLE, BH, KMPH, v.b.) varl nda,
3. Üst ekstremite trombozlar (torasik outlet sendromu veya kateter ile ili kili), retinal ven t kan kl klar nda,
4. Hastanede medikal tedavi amac yla yatan hastalarda,
5. Dü ük riskli, nadir homozigot veya bile ik heterozigot mutasyonlarda aile taramas önerilmez.
6. Arteryel trombozu olan hastalarda, 7. 60 ya üstü hastalarda,
8. Felç geçiren çocuklarda,
9. 60 ya üzerindeki aile bireylerinde kal tsal trombofili taramas önerilmez (33, 34).
9 2.2.4. Kal tsal Trombofilinin Klinik Önemi
Venöz tromboemboli riskini art rmas nedeniyle kal tsal trombofili klinik öneme sahiptir. Kal tsal trombofililerin hemen hepsi otozomal dominant geçi göstermektedir, bundan dolay kal tsal defektleri ta yan hastalar n aileleri de venöz tromboemboli riskine maruz kalabilirler. Hastalar genellikle heterozigottur. Heterozigot PT20210 veya FVL mutasyonlar n trombotik riskte önemsenmeyecek düzeyde art a neden oldu u gösterilmi tir Homozigot hastalarda ise venöz tromboz geli me riski daha yüksektir
Kal tsal etkenlerin varl hastada ve sa kl bireyde artm VTE riskini akla getiriyorsa da, çal malar göstermi ki tek ba na trombofilinin tromboza neden olamayaca ; trombozun birçok kal tsal ve çevresel etkenin bir araya gelmesiyle olu tu unu ortaya koymaktad r (35).
2.2.5. Edinsel Tromboz Nedenleri leri ya
Vasküler Hastal klar Diyabetes mellitus Atheroskleroz
Vaskülitler ( Behçet hastal )
Prostetik materyal (vasküler greftler, kapaklar, santral venöz kateterler, polipropilen, polyester gibi prostetik materyaller)
Trombosit disfonksiyonu Myeloproliferatif hastal klar
Paroksismal noktürnal hemoglobinüri
Anormal reoloji
Hipervizkozite (polistemia vera, Waldenström makroglobüninemisi, akut lökoz, lökostaz, orak hücre hastal , multiple myelom )
10 Hiperkoagülabilitenin e lik etti i di er hastal klar
Oral kontraseptifler, östrojen tedavisi Gebelik
Nefrotik sendrom
Protrombin kompleks konsantrelerinin infüzyonu Trombotik trombositopenik purpura
nflamatuvar ba rsak hastal klar Maligniteler (Trousseau sendromu) Travma
Radyoterapi
FVII yüksekli i, von Willebrand faktör yüksekli i
laçlar (L-asparaginaz, mitomisin, volüm artt s lar, vs.) Antifosfolipit antikor sendromu
Yayg n damar içi p ht la mas
Heparine ba trombositopeni/trombozis (7, 34, 36).
2.2.6. Kal tsal Tromboz Nedenleri
Tekrarlayan trombus ataklar olan veya genç hastalarda meydana gelen trombuslar ile anormal yerle imli trombuslu hastalarda zeminde yatan kal tsal bir hastal k olabilece ini dü ündürmü tür. Kal tsal trombofili nedenleri olarak ilk tan mlananlar; antitrombin III, disfibrinojenemi, protein C ve S eksiklikleridir. Antitrombin eksikli inin ilk kez 1965 y nda tromboza egilim olu turdu u gösterilmi olup, sonras nda 1981’de Protein C eksikligi ve 1984’de protein S eksikliklerinin tromboza e ilim olu turdu u gösterilmi tir. Owen taraf ndan 1947 nda faktör V eksikli inin tan mlanmas , Dahlback ve arkada lar n APC (aktif protein C direnci) 1993’te ve Bertina’n n faktör V Leiden mutasyonunu (G1691A) 1994’te tan mlamalar ile trombozlu hastalar n %20’sinin etiyolojisi ayd nlanm r. 1994 y nda hiperhomosisteineminin, 1996’da Poort ve arkada lar da protrombin genindeki bir mutasyonun (protrombin 20210) tromboza e ilim olu turdu unu göstermi lerdir. Bir hastada birden fazla kal tsal risk faktörünün birlikteli i de olabilmekte, bu durumda tromboz riski daha da artmaktad r. Trombofili aç ndan Hiperhomosisteinemi de önemli bir risk faktörü olarak bilinmektedir. Homosistein
11 biyosentezinde yer alan metilentetrahidrofolat redüktaz (MTHFR) enzimindeki nokta mutasyonlar n (677 ve1298. nükleotidlerde) venöz tromboza yatk nl k ili kisi daha önce yap lan çal ma ve metaanalizlerde de gösterilmi tir (37,38) (Tablo 1).
Tüm bu saptamalara ra men; günümüzde kal tsal tromboz oldu u düsünülen vakalar n %40- 60’n n nedenini ortaya koymak, bütün ara rmalara ra men hala mümkün olamamaktad r. Kal tsal venöz trombozda; ço unlukla p ht la ma sistemini kontrol alt nda tutan do al inhibitör sistem bozukluklar n rol oynad saptanm r. Saptana bu risk faktörlerinden en iyi tan mlananlar ; faktör V Leiden (APC direnci), protein C, protein S, protrombin G20210A mutasyonu, ve antitrombin III eksiklikleridir. lk trombotik atakla ba vuran hastalar n yakla k yar nda; bu eksikliklerden birinin, altta yatan etken oldu u gösterilebilmektedir. Bunlar n nda; hiperhomosisteinemi ve FVIII yüksekli i de önemli risk faktörlerindendir (10, 14, 39, 40).
Tablo 1: Kal tsal trombofili görülme s kl ve trombozla ili kisi (41) Tromboz riskinde art VTE’li hastalarda görülme s kl Normal toplumda görülme s kl (%) FV Leiden heterozigot FV Leiden homozigot 3-8x 50-80x 25–50 5.0 Protrombin 20210A 3x 6 2.0 ATIII eksikli i 25-50x 1 0.02 Protein S eksikli i 10 3 2.0 Protein C eksikli i 10x-15 3 0.3
Disfibrinojemi De ken Dü ük Nadir
12 Tablo 2: Nadir rastlanan primer trombofili nedenleri (41)
Plazminojen aktivatör inhibitör düzeyi yüksekligi. FXII eksikligi
Doku plazminojen aktivatör eksikligi Trombomodulin gen mutasyonlar Disfibrinojemi
Histidinden zengin glikoprotein eksikligi veya yüksekligi Heparin kofaktör II eksikligi
Hipo-displazminojemi (2) Faktör XIII gen polimorphizimi
Faktör I, II, VII, IX, XI düzey yükseklikleri Doku faktör yolu inhibitör (TFPI) eksikli i
Trombinin aktive etti i fibrinöz inhibitörünün (TAFI) yüksekli i Hipofibrinoliz
Tablo 3: Sekonder trombofili nedenleri (41)
Arteriyal tromboz nedenleri Venöz tromboz nedenleri leri ya Ateroskleroz Sigara içme Hipertansiyon Diabetes mellitus Atrial fibrilasyon Sol kalp yetersizli i Oral kontraseptif kullan Östrojen kullan
LDL kolesterol yüksekli i Hipertrigliseridemi Vaskülitik sendromlar
Trombotik trombositopenik purpura, Hemolitik üremik sendrom
Yayg n damar içi p ht la ma sendromu Lipoprotein(a) yüksekli i Antifosfolipid sendromu Polistemi Hipervizkozite sendromlar Lökostazis sendromlar leri ya Travma mmobilizasyon Genel cerrahi giri im Ortopedik cerrahi giri im Konjestif kalp yetersizli i Nefrotik sendrom
Oral kontraseptif kullan Östrojen kullan Obezite Gebelik Postpartum dönem Varisler Malignite Behçet Hastal Antifosfolipid sendromu
13 2.2.6.1. Antitrombin III Eksikli i
Antitrombin; 432 aminoasitten olu an tek zincirli bir plazma glikoproteini olup, 58.000 dalton a rl ndad r. Karaci erde ve Endotel hücrelerinde sentezlenir. Serin proteaz inhibitörleri ailesinden olup, primer trombin inhibitörüdür, ayr ca di er serin proteazlar da (FIXa, FXa, FXIa, FXIIa ve kallikrein) inhibe eder. Haliyle fibrin formasyonunun en güçlü fizyolojik inhibitörüdür. Heparin veya heparin benzeri moleküllerin varl nda, etkisi yakla k 1000 kat artar.
Toplumda genel s kl 1/600–1/5000 olarak tahmin edilmektedir. ATIII eksikli i kal tsal trombofilik hastal klar n en güçlü trombojenik olan r ve hastalar ya amlar boyu %50’den fazla oranda tromboembolik olay geçirme riski alt ndad r (31). Venöz tromboembolilerin %2 ile %6’s ndan sorumludur. Otozomal dominant geçi gösterir. ATIII eksikli i olanlar n %85’ inde 50 ya na kadar tromboemboli gözlenir (32). ATIII eksikli i hetorojen bir bozukluktur ve otozomal dominant olarak geçen 80’den fazla mutasyon bildirilmi tir. ATIII eksikli inin; genel populasyonda kl %0.02, venöz trombozlularda %1 olup tromboz riskini 50 kat artt rmaktad r. Homozigot antitrombin eksikli i bildirilmemi olup muhtemelen hayatla ba da mamaktad r.
ATIII eksikli inde arteriyel tromboembolik olaylar karakteristik de ildir. En çok etkilenen venler iliofemoral ve femoropopliteal venlerdir. Di er venlerde de VTE olaylar görülebilmektedir. Hastalar n %60’ nda tekrarlayan VTE, %40’ nda ise pulmoner tromboemboliye (PTE) rastlanmaktad r. Ya la birlikte tromboz görülme
kl giderek artmaktad r. ATIII eksikli i olan baz ki ilerde heparin tedavisine direnç görülür. ATIII eksikli i özellikle gebelikte VTE olaylar aç ndan belirgin risk art na yol açmaktad r (2, 42, 43).
Gende meydana gelen farkl mutasyonlar sonucu, Tip I ve Tip II ATIII eksikli i olarak 2 gruba ayr r.
Tip I AT eksikli i: Hastalar n çogu heterozigottur ve ATIII miktar azalm r ve aktivitesini gösteren fonksionel testlerde bozulmu tur.
Tip II AT eksikli i (Varyant AT molekülü): Nokta mutasyonlar sonucu olu ur. AT molekülünün serum düzeyi normaldir ancak fonksiyonu yetersizdir. Hastalar n antitrombin aktivite düzeyleri dü ük fakat antitrombin antijenleri normaldir.
14 a) Tip II RS (reaktif bölge) mutasyonlar : Antijen miktar normal, ancak aktivite dü üktür. AT molekülünün, trombinle birle me bölgesinde de ikli e neden olurlar.
b) Tip II HBS (heparin ba lanma bölgesi) mutasyonlar : AT molekülünün, heparin ba lanma bölgesinde de ikli e neden olan mutasyonlard r. AT’nin heparine ba lanmas bozuk oldu undan, heparine direnç görülür.
c) Tip II PE (pleiotropik etki) mutasyonlar : hem reaktif bölgeyi Hem de heparin ba lanma bölgesini etkileyen mutasyonlard r.
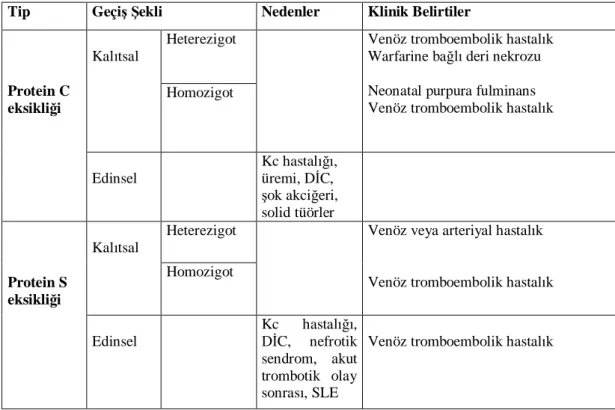
2.2.6.2. Protein C Eksikli i
Protein C, 62000 D a rl nda bir serin proteaz olup, hafif ve a r zincir olmak üzere 2 polipeptid zincirinden olu ur ve 406 aminoasit içerir (44). PC geni, 2. kromozomda 2q13-2q14 bölgesinde lokalizedir. Sentezi karaci erde yap lan K vitaminine ba ml bir protein olan PC geninde bugüne kadar 160 farkl mutasyon tan mlanm r (45, 46).
Kal genellikle heterezigotlarda otozomal dominant olup, homozigotlarda otozomal resesiftir. Toplumdaki s kl 1/200–1/300 olarak bildirilmi tir. tromboza rastlanma oran 50’li ya larda en s k görülür, 20 ya ndan önce dü üktür. Genellikle spontan tromboza neden olurken %30 uyar bir faktör katk da bulunur. En s k alt ekstremite venlerinde tromboza neden olurken, atreriyel trombozada neden olabilir.
Edinsel protein C eksikliklerinin sebebi ise oral antikoagülan kullan , yayg n damar içi p ht la ma (D C), karaci er yetmezli i, antifosfolipid sendromu, üremi, immobilizasyon, ok akci eri ve postop solid tümörler olarak say labilir (Tablo 5).
Klinikte 2 tip protein C eksikli i tan mlanmaktad r; (47, 48)
Tip I PC eksikli i: Proteinin moleküler yap normal olup, antijeni ve aktivitesi azalm r. Buna CRM (-) tip ad da verilmektedir. Delesyon, yanl anlaml ve anlams z mutasyonlar tip I PC eksikli i nedeni olarak bildirilmi tir (46, 49). Tip II PC eksikligi: Antijenik PC miktar normal, fakat bu sefer de molekül bozuk ve i levsizdir. Buna CRM(+) tip de denilmektedir (46, 50, 51). Bu eksikli e neden olan mutasyonlar, yanl anlaml mutasyonlard r. Toplumda s kl %0.02
15 olan, PC eksikli i hastalar n ço u tip I heterozigottur ve klini i çok de ken olup kal tsal PC eksikli i tan koymak oldukça güçtür (46, 52, 53).
2.2.6.3. Protein S Eksikli i
PS eksikli inin; homozigot ekli otozomal resesif iken, heterozigot ekli kal otozomal dominant kal r. Genel populasyonda s kl 1/1000 olup, klinikte 3 tip PS eksikli i tan mlanm r.
Tip I PS eksikli i: Hastal n en s k ekli olan tip I PS eksikli inde; total PS antijeni, serbest PS antijeni ve PS aktivitesi birlikte azalm r. Tip I PS eksikli ine neden olan mutasyonlar n ço u nükleotid degi imi, delesyon ve insersiyonlard r.
Tip II PS eksikli i: Bunlarda serbest PS düzeyi dü ük, total PS düzeyi normaldir. Bu tip eksikli in nedeni olarak 1’i k rp lma bölgesi, 5’i yanl anlaml mutasyon olmak üzere toplam 6 mutasyon bildirilmi tir
Tip III PS eksikligi: Bu eksikli e ise; yaln zca PS aktivitesi dü üktür 9’u anlaml , 1’i nükleotid de imi olmak üzere 10 mutasyonun neden oldu u bildirilmi tir. S kl venöz trombozlularda %1–2 olan PS eksikli inin, tromboz riskini yakla k 10 kat artt rd dü ünülür (Tablo 4).
Tablo 4: Protein C ve protein S eksikli i tipleri, ekilleri ve klinik tipleri
Tip Geçi ekli Nedenler Klinik Belirtiler
Heterezigot Kal tsal
Homozigot
Venöz tromboembolik hastal k Warfarine ba deri nekrozu Neonatal purpura fulminans Venöz tromboembolik hastal k Protein C eksikli i Edinsel Kc hastal , üremi, D C, ok akci eri, solid tüörler Heterezigot Kal tsal Homozigot
Venöz veya arteriyal hastal k
Venöz tromboembolik hastal k Protein S eksikli i Edinsel Kc hastal , C, nefrotik sendrom, akut trombotik olay sonras , SLE
16 2.2.6.4. Aktif Protein C (APC) Direnci ve Faktör V Leiden Mutasyonu Genetik olarak kar za ç kan en s k trombofili nedenidir. Venöz tromboembolilere neden olur. Normal artlarda plazmaya APC eklendi inde FVa ve FVIIIa’n n parçalanmas sonucu aktive parsiyel tromboplastin zaman (aPTT) uzar. Dahlback ve arkada lar 1993 y nda üç hastan n plazmas na APC eklenmesiyle sonucu aktive parsiyel tromboplastin zaman nda (aPTT) beklenen uzaman n çok az oldu unu veya hiç olmad bildirmi lerdir. Bu durumu APC direnci olarak tan mlam lard r. Dahlback APCR’na, FV ya da FVIII genlerindeki bir bozuklu a veya PC aktivasyonunda rol oynayan bir kofaktörün eksikli ine ba olabilece ini öne sürmü tür (54).
Sonraki çal malarda ise vakalar n plazmas na FV eklendi inde APCR’n n olmad görülmü tür (55). 1994 y nda, APCR’n n moleküler patolojisi Bertina ve arkada lar taraf ndan aç klanm r. Birinci kromozomun uzun kolu üzerinde bulunan FV geninin 1691. pozisyonundaki adenin ile guaninin yer de tirmesi sonucu ortaya ç kan mutant gen, FV’in a r zincirinin 506. pozisyonundaki arginin yerine glutamin geçmesine neden olur. Bu FV molekülünü Faktör V Leiden (FVL) olarak tan mlam lard r. APCR’n n en önemli nedeni, FV geninde tek nokta mutasyonu olarak bildirilmi tir (2, 56).
FV proteini; 2196 aminoasitten olu mu , 330.000 D moleküler a rl nda tek zincirli bir glikoproteindir. karaci er ve megakaryositlerde sentezlenir. FV proteini, koagülasyon mekanizmas n hem ekstrinsik hem de intrinsik yollar nda görev yapan bir kofaktördür. FVa, FXa ve trombosit fosfolipidleri ile birle erek protrombin aktivatörü kompleksini olu turur; olu an bu kompleks protrombinin (FII), trombine (FIIa) dönü ümünü gerçekle tirir. Trombinin, fibrinojeni fibrine dönü türmesi sonucunda p ht olu ur. P ht la ma ba lay nca trombin; tek zincirli ve inaktif FV molekülünü belirli noktalardan keserek, birbirlerine Ca++ iyonlar ile ba çift zincirli aktif molekül haline dönü türür. Aktive olmu FV (FVa), protrombin aktivasyon h yakla k 2000 kat artt r (2,57).
APC, normal artlarda FVa’y 3 yerinden, Arginin 306, 506 ve 679 noktalar ndan parçalar. 506. noktada arginin yerine glutamin geçmesi sonucu FVa, APC’nin etkisine kar dirençli duruma geçer (58). APCR protein C yolundaki pek çok anomaliye ba olabilir. Bu anomaliler defektif APC substratlar ü defektif APC
17 kofaktörleri veya normal bir protein C yoluna kar olu mu antikor veya di er ajanlardan kaynakl olabilir. APC’ye kar geli en direnç otozomal dominant olarak kal lmaktad r (59). P ht la ma kaskad n aktivle mesiyle ortaya ç kan trombin bir taraftan fibrinojeni fibrine dönü türürken, di er taraftan trombomoduline ba lan r. Bu olay trombinin prokoagulan durumdan antikoagulan duruma dönü mesine yol açar. Sonras nda trombin, PC’yi aktive eder. APC, kofaktör PS varl nda FVa’y önce 506. pozisyondan, sonra s ras yla 306. ve 679. pozisyondan olmak üzere; FVIIIa’y da özgül bölgelerden keserek inaktive eder. FVa ilk olarak 506. pozisyondan kesildi inde FVa olu ur. Bu form, %70 oran nda prokoagulan aktiviteye sahiptir. FVa’n n tam inaktivasyonu için 306. pozisyondan da kesilmesi gerekir. Bu reaksiyon PS taraf ndan 20 kat artt r, bu reaksiyonu artt etkiye sahiptir (59).
Sonradan edinilmi APCR, genelde asemptomatik olup; oral kontraseptif kullan , hamilelik ve cerrahi müdahaleler gibi baz edinsel faktörlerin e li inde, tromboz geli imine neden olur (60). FVL mutasyonu, ço unlukla alt extremitelerde DVT ve e er emboli geli iyorsa, PTE olarak kendini gösteren venöz tromboembolizmde s k rastlanan kal tsal bir faktördür.
FV geninde meydana gelen nokta mutasyonu sonucu mutant FV, normale göre on kat daha yava inaktive olup, dola mda daha uzun süre kalmaktad r. Bu durum daha fazla trombin üretimine ve protrombin fragmanlar ndan FXII’nin ve aktive olmu koagülasyon faktörlerinin art na sebep olarak hiperkoagulopatiye neden olmaktad r. Bu durum, FV molekülüne, APC’nin proteolitik etkisine kar direnç kazand rmakta ve neticede bu mutasyonu ta yan bireyler venöz tromboza ilimli hale gelirler. Bu mutasyon, heterozigot ve homozigot olarak vücutta ta nabilir. Homozigot FVL mutasyonu olan bireyler, heterozigot mutasyonu olan bireylere göre daha fazla tromboz riski ta maktad r; bu durum heterozigot olanlarda normal FV’in de bulunmas ve bu sayede APC’nin FVIIIa’y inaktive ederek antikoagulan etki sa lamas yla aç klanabilir (61).
Homozigot FVL mutasyonu olanlarda Tromboz riski 50–100 kat. Heterozigot mutasyonu ta yanlarda ise tromboz riski 5–10 kat artmaktad r. Genel popülasyonda heterozigot FVL mutasyonu %3–7 oran nda saptan r. FVL mutasyonu olanlarda 2 kat oran nda trombüs nüksü fazlad r (62).
18 2.2.6.5. Protrombin G20210A Mutasyonu
Protrombin (FII), koagülasyon kaskad n en son basama nda trombinin prekürsörü olup, 11. kromozomun k sa kolunda bulunur. Karaci erde (KC) sentezlenen K vitamini ba ml bir faktördür. Protrombin molekülü; 71.000 D rl nda, 579 aminoasit içeren ve ag rl n yaklas k % 10’u karbonhidrattan olusan tek zincirli glikoprotein bir yap r. PTGM, Poort ve arkada lar taraf ndan 1996 y nda; protrombin geninin 20210. nükleotidde, guanin yerine adenin gelmesiyle meydana gelen tek baz de im mutasyonu (G20210A) tan mlanm r. Olu an bu mutasyon, mRNA degradasyonunda yava lamaya yol açarak serum protrombin düzeyini artt rmak suretiyle koagülasyonda art a yol açmaktad r (63). Dünyada yakla k %3 s kl kta bulunan bu mutasyonun VTE bulunan hastalarda % 4 – 17 aras nda oldu u gösterilmi tir. Heterozigot ta larda, normal bireylere göre protrombin seviyesinin %30 daha fazla oldu u gösterilmi tir (12).
Heterozigot PTGM ta lar nda, serebral ven trombozu ve DVT riski artt gibi, rekürren VTE oran nda da art saptanm r. Geli en bu mutasyonun, venöz tromboemboli riskini 3 kat, baz ara rmalara göre 8 kat artt rd gösterilmi tir. lave bir kal tsal risk faktörünün varl nda, özellikle FVL mutasyonu ile birlikte, VTE riski yakla k 20 kat yükselmektedir. Bu mutasyonun, gebelik s ras nda geli en tromboembolilerde de rolü oldu u gösterilmi tir. Trombofilik olaylar ile plasentan n perfüzyonu bozulmakta ve spontan abortus geli mesine neden olmakta, intrauterin geli me anomalilerine neden olarak preeklempsiyede neden olabilmektedir. Mutasyonunun di er genetik tromboz yap faktörler ya da kolayla faktörlele birle ti inde risk olu turdu u saptanm r. Özellikle VTE’li genç hastalar ve tekrarlayan tromboembolisi olan ya hastalarda bak lmas önerilmektedir (64, 65).
2.2.6.6. Hiperhomosisteinemi
Bir metionin metabolizma bozuklu u olan hiperhomosisteinemi; hem venöz hem de arteriyel tromboza neden olabildi i gösterilmi tek kal tsal risk faktörüdür. D’Angelo ve Selhub taraf ndan 1997’de arteriyal tromboz için risk faktörü olarak tan mlanm , 1994’te Falcon, 1995’te Fermo taraf ndan tromboembolizm, Heijer taraf ndan da 1996 y nda derin ven tromboz ve tekrarlayan venöz trombozu için kal tsal bir risk faktörü oldu u tan mlanm r.
19 Homosistein, 5-metil tetrahidrofolat varl nda, B12’ye ba ml metiyonin sentaz (MS) taraf ndan metiyonine dönü mektedir. Sonras nda, 5,10-metilen tetrahidrofolat, MTHFR ile 5-metil tetrahidrofolata indirgenir. Betain-homosistein metil transferaz arac yla karaci er ve böbrekte homosisteinin yeniden metilasyonu, gerçekle mektedir. Beslenme ile al nan metiyonin, metiyonin siklusunda metil transferaz ile, metil grubu vericisi olarak i gören S-adenozil metiyonin (SAM)’e dönü mektedir. Bu reaksiyonda olu an di er bir ürün, S-adenozil homosistein hidrolaz taraf ndan, homosistein ve adenozine hidrolizlenen S-adenozil homosistein (SAH)’dir. Homosistein, katabolik transsülfürasyon yoluna da girmektedir. Bu yoldaki ilk enzim, B6 vitaminine ba ml sistatiyonin -sentaz (CßS)’d r. Sistatyonin, B6 vitaminine ba ml sistatiyonaz arac yla sisteine dönü mektedir (66).
Hücre içi homosistein metabolizmas üç enzim; metiltetrahidroflat redüktaz (MTHFR), sistathionin beta sentetaz ve metionin sentetaz ile enzimatik olaylarda kofaktör rolü oynayan üç vitamin (B12, folat, B6) içerir. Bu enzimlerdeki defekt ve kal tsal eksiklik homosistein düzeylerinde yüksekli e ve folat düzeylerinde azalmaya yol açarak klinikte prematür arteriyel vasküler hastal k ile venöz ve arteriyel tromboza neden olur. Bu enzimlerdeki kal tsal bozukluklar nadir olup, hiperhomosisteinemi nedeni olarak daha çok edinsel vitaminlerin (B12, folat, B6) eksikli i söz konusudur. B6, B12 vitaminleri ve folik asit aç ndan yetersiz bir beslenme, hiperhomosisteinemi nedenidir.
Açl k plazma homosistein konsantrasyonu normalde 5–15 µmol/L iken, hafif hiperhomosisteinemide 15–30 µmol/L, orta hiperhomosisteinemide 30–100 µmol/L ve ciddi formunda ise 100 µmol/L’den fazlad r. Genel populasyonda % 5-10 oran nda homosistein oran yüksek bulunurken, trombozda % 25’lere kadar ç kmakta ve tromboz riskini 3-4 kat artt rmaktad r (1, 2, 66, 67).
2.2.6.7. Faktör VIII Yüksekli i
FVIII; 2332 aminoasitli, 265 kD a rl nda oldukça büyük tek zincirli glikoprotein yap da olup; intrinsik p ht la ma yolundaki FX’un aktivasyonunda görevlidir ve FIXa’n n kofaktörüdür.
20 FVIII’i kodlayan en az 3 gen oldu u bilinmektedir ve çok say da lokus taraf ndan p ht la ma faktörlerinin konsantrasyonlar etkilemektedir. Bu genlerden biri ABO kan grubunu kodlayan gendir ve 0 grubu bireylerde FVIII ve vWF düzeyi daha dü ük konsantrasyondad r. 0 kan grubu haricindeki kan gruplar n tromboz riski ile ba lant oldu u gözlenmi tir (68, 70).
Venöz trombozlu karde lerde artm FVIII düzeyi ve bu yüksekli in sebat ediyor olmas faktör VIII’in herediter bir yatk nl k sa lad ve ba ms z bir risk faktörü olu turdu unu dü ündürmektedir. Baz çal malarda FVIIIC aktivitesinde her bir 0,1U/L art ta, venöz tromboz riskinin % 10 artt bildirilmektedir. FVIII yüksekli i tromboz risk art nda ba ms z bir faktördür ve rekürrens ile ili kilidir. Trombozu olan hastalarda FVIII düzeyinin ölçülmesi büyük yarar sa lar.
2.2.6.8. Fibrinojen Gen Mutasyonu
Fibrinojen molekülü bir plazma proteinidir ve alt polipeptit zinciri içerir ki bu zincirler 2 (alfa), 2 (beta) ve 2 (gama) zinciridir. Fibrinojen molekülünün uçlar yüksek derecede negatif olarak yüklüdürler bu özelli i, suda çözünürlü e katk da bulunur ve di er fibrinojen molekülleri ile birbirlerini iterek agregasyonu önler. Fibrinojeninin plazmada normal de er aral 200-400 mg/dl kadard r (68, 69) ve karaci erde sentezlenir. Kan n p ht la mas , solubl plazma proteini fibrinojenin (faktör I), insolubl polimerlerinin fibröz a haline enzimatik dönü ümüdür. Fibrinojenin (faktör I) dönü ümünde rol alan enzim, trombin (faktör IIa)’dir (69, 70).
Fibrinojenin plazma seviyeleri cinsiyet, ya , genetik ve hormonal faktörler (obezite, diabetes mellitus, hiperkolesterolemi) sigara içimi ve oral kontraseptif kullan gibi fiziksel etmenlerle de mektedir, ayn zamanda akut faz reaktan olup infeksiyon ve so uk gibi enflamatuar durumlarda da plazma düzeyi artmaktad r.
Fibrinojen kan düzeylerinde art a yol açan genetik bozukluklar içerisinde en s k subünitiyle ilgili gen polimorfizmleri bulunmaktad r. -Fibrinojen gen polimorfizmiyle ili kili olarak yakla k 10 mutasyon tan mlanm olmakla birlikte vask ler patolojide rol daha iyi anla lm olan -Fibrinojen -455G/A ( Hae III) gen polimorfizmidir. Plazma düzeyi yüksek saptanan fibrinojen endotel hücreleri ve subendotelyal kollajen kümele mesiyle birliktedir. Trombosit
21 agregasyonun art na neden olup di er risk faktörlerinin birlikteli inde tromboza
ilimi artt rmaktad r (71, 72).
2.2.6.9. Plazminojen Aktivatör nhibitör 1(PAI–1) Gen Poliformizmi Homeostaz n düzenlenmesinde fibrinolitik sistemde iki enzim sistemi önemli rol oynamaktad r. Bu enzim sistemlerinden birincisinde, plazminojen aktivatörleri, ikincisinde ise plazminojen aktivatör inhibitörleri yer almaktad r. Plazminojen aktivatörleri aras nda doku plazminojen aktivatörü (t-PA) ve ürokinaz, plazminojen aktivatör inhibitörleri aras nda ise plazminojen aktivatör inhibitör–1 (PAI–1) vard r. Fibrinolitik sistemde PAI–1 aktivasyonu fibrin taraf ndan gerçekle tirilmekte ve aktive olan PAI–1 ise t-PA'y ba layarak onun litik etkisini inhibe etmektedir (73).
PA –1 serpin ailesine ait bir serin proteinaz inhibitörüdür. Karaci er ve endotelyal hücrelerde sentez edilir. Az miktarlarda olmas na ra men plasmada saptanan PA –1 yüksek aktiviteye sahiptir. plateletlerde yüksek oanda bulunmas na ra men dü ük aktiviteye sahiptir (74, 75).
PA -1’in cinsiyet, obesite, ya , plasma tumor ntrlökin–1 (IL–1), tumor growth faktor (TGF- ), nekrozis faktor (TNF- ) konsantrasyonu, lipopolisakkarid, phorbol esterler, glukokortikoid hormonlar, VLDL ve insülinden etkilenmektedir (76, 77). Bir sirkadiyen ritme sahiptir ve sabah erken saatlerde pik yap p ilerleyen saatlerde tekrar dü er PA –1 tromboembolik olaylarla ili kili olan di er sitokin ve hormonlarla benzerlik göstermektedir. PA –1 bir akut faz reaktan olarak tan mlanmaktad r. Cerrahi sonras , Miyokard enfarktüsü, endotoksemi ve septisemi gibi durumlarda artm PA –1 aktivitesi artar.
PA –1’in yar ömrü 4 saaten azd r. Vitronektin PA -1 kofaktorü gibi hareket eder, ve yar ömrünü yakla k on dakika içinde iki ile üç kat artt rarak PA -1 ‘i aktif formda stabilize eder (75, 78, 79). PA –1 molekülü h zl bir ekilde doku plasminogen aktivatörü (tPA) ve uroplasminogen akt vatorü(uPA) ile birlikte hareket eder ve kompleks olu turur. PA –1 kanda dü ük konsantrasyonlarda bulunmas na ra men doku plasminojen aktivatörü (tPA) ve uroplasminojen aktivatörünün (uPA) invivo inhibisyonundan sorumludur.
Yak n zamanda PA –1 geninde sekiz polimorfizm ke fedildi. Ancak bu polimorfizmlerden sadece bir kaç PA –1 sentezi, plasma konsantrasyonu ve
22 aktivitesi üzerinde etkili görünmektedir (80). PA–1 promotor genotip aç ndan homozigot 4G/4G ve heterozigot 4G/5G polimorfizmleri genellikle daha yayg n bulunmaktad r. Bununla birlikte, 4G alleli sitokin sitimulasyonu alt nda yüksek plasma PA –1 promotor aktivitesi ile ili kili oldu u bulunmu tur.4G alleli için homozigot(4G/4G homozigot mutant ) olan ki ilerde plazma PAI–1 konsantrasyonun 5G için homozigot (5G/5G homozigot normal) olan bireylere göre % 25 oran nda daha fazla olabilece i gösterilmi tir (81).
Genetik ve çevresel faktörlerin etkisi alt nda, PA –1 yüksekli i birçok medikal durumda arteriyel ve venöz trrombüs formasyonunun geli imi ile ili kilendirilmi tir.
2.2.6.10. Faktör XIII Gen Polimorfizimi
Fibrin stabilizatör faktör olarak bilinen Faktör XIII’ün hemostazdaki as l rolü fibrin zincirlerine çapraz baglanmas ve fibrinolizisin regülasyonunda önemli proteinlere kovalent olarak yap mas r. Fibrin zincirleri çapraz baglamas yla p ht stabilizasyonunda önemli bir rol oynar.
FXIII bir transglutaminazd r A ve iki B alt birimlerinden olusur. Plazmadaki dolas tetramer yap dad r (A2B2). Dolas mdaki FXIII’ün %90’ ndan fazlas B alt birimi bir baglay kenar yla fibrinojene baglan r. Bu etkilesim durumundan fibrinojen plazma zimojen için bir tas gibi hizmet eder (82).
FXIII’ün aktivasyonun olusumu trombin taraf ndan 37 aminoasitten olusan bir aktivasyon peptidinin ayr lmas yla olur. Olusan aktif faktör XIII biti ik fibrin molekülleri aras nda gama-glutamil epsilon-lisil baglar çapraz baglar. Fibrinin intermoleküler çapraz baglanmas fibrinolize direnci ve p ht stabilitesini art r (83). Ayr ca antiplazminin çapraz baglanmas fibrinolize direnci daha da art r. Alfa–2 antiplazmin, vWF, FVII, kollajen, fibronekin, trombospandin ve vitronektin FXIII için gerekli substrat proteinlerdir.
FXIII ile ilgili birçok genetik polimorfizmi tan mlanm r (84). Daha önceki çal malarda en çok FXIII A subünitesindeki genetik polimorfizm ara lm r. FXIIIA subünitindeki genetik polimorfizmi toplumlar aras da büyük farkl k göstermektedir. Lösin 34 allel s kl bat toplumlar nda daha yüksek oranda bulunmu tur. FXIIIA subünitindeki genetik farkl k FXIII’ ün plazma seviyesini ve
23 FXIII aktivitesini etkileyebilmektedir (85, 86). Lösin varyant FXIII’ ün transglutaminaz aktivitesini de tirmekte; daha yüksek enzim aktivitesine neden olmaktad r.
Heterozigot ta larda orta derecede enzim aktivitesi ile karakterizedir (85). Plazma FXIII art yeterli miktarda plazmin olu umunu engelleyerek daha rigit yap da fibrin olu turmakta, fibrinin fibrinolitik etkilere kar daha dirençli olmas na neden olmaktad r. Lösin 34 varyant n MI riskini art rd gösteren çal malarda farkl sonuçlar elde edilmi tir (86).
2.2.7. Atipik Trombozlar
Alt ekstremiteler d nda venöz tromboz; üst eksremite derin venleri, serebral sinüsleri, retinal venler ve abdominal venler gibi vasküler alanlardan meydana gelebilir. VTE’nin bir ba ka nadir görülme ekli, objektif tekniklerle tan mlanamayan emboli kayna olan ‘’izole’’pulmoner embolizmdir. Atipik lokalizasyonda ven trombozu, tekrarlayan ven trombozu, genç hastada tromboz, aile öyküsünün pozitif olmas gibi durumlarda tromboz etyolojisini ara rmak için kal tsal ve edinsel risk faktörleri detayl olarak ara lmal r. Al lmad k, s k görülmeyen bölgelerdeki venöz trombozlar hakk nda iyi kay tlar mevcuttur (iyi dökümante edilmi lerdir) ancak bu bölgelerde görülen venöz trombozlar ile ilgili çal ma yürütmek zor ve s nt olmu tur. Bu nedenle bu bölgelerde görülen venöz trombozlar n yönetimi ve tedavisi s kl kla alt ekstremite DVT (derin ven trombozu) ve PE (pulmoner emboli) yönetiminden esinlenilerek (tahmini olarak –benzer
ekilde) düzenlenmi tir.
2.2.7.1.Serebral ve Sinüs Ven Trombozlar
Serebral ven ve sinüs trombozlar n (CVST) tüm inmelerin %1’inden daha az ndan sorumludur ve daha çok genç-eri kin ve çocuklar (bunlar n da %75’i kad nd r) etkilemektedir (87). Y ll k insidans raporlar na göre her y l toplumda 4 milyonda bir, 7 milyon çocukta bir ve 12 milyon do umda bir CVST görülmektedir.
nfeksiyon (özellikle ba ve boyun enfeksiyonlar ), sistemik inflamatuar bozukluklar, lösemi (özellikle aspariganase-asparajinaz tedavisi alanlar), kafa travmas ve dehidratasyon özellikle tan mlanm allta yatan sebeplerdendir. Polisitemia vera ve
24 esansiyel trombositemi gibi myeloproliferatif kanserlerin (MPNs) seyri esnas nda CVST görülebilir(88) .Östrojen-kombine oral kontraseptiflerin (C-OCPs) kullan da presipite edici bir faktördür(89) (Bu hastalar a daki mutasyon yada eksiklikleri ta yabilirler F5 R506Q (factor V Leiden mutasyonu) ile birlikte F2 G20210A (prothrombin gene mutation) ve h zl yükselen homosistein seviyeleri ile birlikteli inde (90). Tedaviyi e er CVST hastada geçici bir risk faktörüne ba ysa bir vitamin K antagonisti ile 3 ay boyunca devam ettirmek gerekir; e er unprovake (sebepsiz-provake eden nedenler yoksa) CVST si mevcutsa veya hafif trombofilisi mevcutsa (örne in heterozigot F5 R506Q veya F2 G20210A mutasyonu varsa) vitamin K antagonistini 6-12 ay boyunca devam etmek gereki (91) Tekrarlayan CVST ataklar geçiren hastalarda ve sadece bir kere CVST ata geçirmi ancak ciddi trombofilisi olan (antitrombin, protein C veya protein S eksikli i varsa, homozigot F5 R506Q veya homozigot F2 G20210A mutasyonu varsa, antifosfolipid antikor varsa veya kombine anomaliler varsa) hastalarda uzun süreli antikoagülasyon uygulanmal r (91). Alternatif bir uzman görü üne göre de unprovake CVST'si olan hastalarda uzun süreli antikoagülan kullan için endikasyonlar öyledir: p ht n tekrarlayan görüntülemelerde tam olarak rezole olmamas (erimemesi), sürekli risk faktörlerine sahip olmak veya trombofili bulunmas r (92). Herediter trombofili testi yap lmas tekrarlayan CVST için net olmayan prediktif bir de ere sahiptir. Ancak bununla birlikte baz uzmanlar antitrombin, protein C ve protein S eksikli i olan hastalarda sürekli antikoagülan tedavi önermektedirler.
2.2.7.2. Üst Extremite Venöz Trombozlar
Aksiller, subklavyan ve brakiyal venlerin trombozu Üst extremite derin ven trombozlar aksiler, subklavyan ve brakiyal venleri içerebilir. Tüm DVT’lerin %10’unu içerir ve y lda 100 bin ki iden 16’s nda meydana gelir (93). Üst extremite derin ven trombozlar idiopatik veya torasik outlet sendromu (TOS) veya paget-schroetter sendromu’na (egzersize ba geli en üst extremite derin ven trombozu) (Paget, 1875; Von-Schroetter, 1884) ba geli iyorsa primer olarak geli mi tir. Santral venöz kateter gibi sekonder bir sebebe ba da geli ebilir. Bu nedenle ilk olarak primer ve sekonder ayr yap lmal r. Santral venöz kateter, aktif malignensi ve kazan lm yada herediter trombofililer Üst extremite derin ven
25 trombozlar için risk faktörü olarak de erlendirilmektedirler. En s k risk faktörü santral venöz kateter uygulanmas r (93). Pacemakerlar da bilinen üst extremite derin ven trombozu risk faktörlerindendir. Alt ekstremite cerrahisi ve alç sonras immobilizasyon üst extremite derin ven trombozlar için s ras yla 13 ve 7 kat daha fazla risk olu turmaktad r (94). Kombine oral kontraseptif kullan ve hormon replasman kullan cidd birer risk faktörü olamam lard r.
2.2.7.3. Juguler ven Trombozu
Juguler Ven Trombozu: Lokal sepsis travma ve inflamasyon ile ba lant olarak s kça görülmektedir. Ço u kez Lemierre Sendromunun bir parças olarak tan mlanm r ki bu sendrom yak n zamanlarda geçirilen bir orofarengeal enfeksiyon ile karakterize, internal juguler ven trombozuna ait klinik ve radyolojik kan tlar n bulundu u ve Fusobacterium necrophorum ba ta olmak üzere anaerobik patojenlerin kl kla sebep oldu u(izole edildi i) bir sendromdur. Overyan hiperstimülasyon sendromu bulunan hastalarda juguler ven trombozu geli imde art oldu u görülmektedir (95). Tedavi ve görüntüleme üzerine veriler s rl r.
2.2.7.4. Vena Cava Trombozu
Superior vena cava trombozu: Süperior vena cava (SVC) obstrüksiyonu malignensi yada benign hastal klardan kaynaklanabilir. Maligniteler %60’l k bir oranla en s k rastlanan SVC trombozu sebebidir (96). Non-malign sebepler aras nda
kl kla; Santral venöz kateter cihazlar yada pacemaker araçlar n (tel-kablo gibi) uygulanmas bulunmaktad r (96). Di er tan mlanm sebepler aras nda enfeksiyon ve mediastinal fibrozis gelmektedir. CT tarama genel olarak ilk uygulanan görüntüleme yöntemidir Kal risk faktörü bar nd ran hastalara sürekli antikoagülasyon uygulanmas de erlendirilmelidir.
2.2.7.5. nferior Vena Cava Trombozu
nferior vena cava trombozu etyolojisi derin ven trombozu ile benzerdir. Malignite, özellikle de renal hücreli karsinom gibi nferior vena cava’ya yak n di er tümörler nferior vena cava trombozuna sebep olabilirler (direk vena cava infeiora invazyon daya bas yapmak suretiyle). nferior vena cava trombozu’nun konjenital
26 anomalisi gibi non-malign risk faktörleri yada external bas gibi sebepler direk olarak venöz staza veya türbülan kan ak na neden olmaktad rlar. nferior vena cava trombozu’lu birçok vaka alt extremite derin ven trombozu (DVT) veya PE(pulmoner emboli) semptom ve bulgular yla prezente olmaktad r. Bilateral alt extremite DVT’leri ile prezente olan hastalara nferior vena cava trombozu görüntülemesi mutlaka yap lmal r. MAISTHRO VTE kay tlar nda hastalar n %10 unda bir lupus antikoagülan bulunmu tur ancak nferior vena cava trombozu geçiren hastalarla, alt extremite DVT geçiren ayn ya ve cinsiyette hastalar ile k yasland nda herediter protrombotik anomaliler çok fazla görülmemi tir.(herediter protrombotik anomaliler alt extremite DVT geçiren hastalarda IVCT geçiren hastalara oranla daha s k görülmü tür) (97). Belirli tiplerde IVC filtreleri kullanan hastalarda yüksek IVCT.
2.2.7.6. Portal Ven Trombozu
Portal ven superior mezenterik ve splenik venlerden olu mu tur. Portal ven trombozunun altta yatan en s k etyolojisi sirozdur (98). Di er sebepler ise; intra-abdominal enfeksiyonlar, inflamasyon yada malignite, künt travma yada cerrahidir. Myeloproliferatif hastal klar (MPN) vakalar n dörtte birini olu turmakta ve PVT ise MPN’nin yayg n prezentasyonlar ndan birisi olarak bulunmaktad (99). Herediter trombofililer etyolojik faktörlerdir ve antifosfolipid antikorlar da muhtemel etyolojik bir sebep olarak görülebilirler.
Portal ven trombozu prezentasyonu akut kar n a , ate ve bulant gibi akut ekilde olabilece i gibi portal hipertansiyon semptomlar ile (varis kanamas , asit, hipersplenizm) kronik de olabilmektedir. Tan CT, Doppler USG ve MRI ilekonulabilmektedir. Antikoagülasyonun devaml (sürekli uygulan p uygulanmamas gerekti ine ba ) konusunda herhangi bir rehber yoktur. Ancak bununla birlikte herediter trombofilisi olan veya rekanalizasyon s nt olan hastalarda uzun süreli antikoagülasyonun uygulanmas gerekti i yönünde görü ler vard r ancak bu görü ler herhangi güçlü delillere dayand lamam r (100-102). MPN ‘si olan hastalarda görülen PVT’yi takiben tekrarlama s kt r (%27-33) (103) ve bu hastalar için uzun süreli antikoagülasyon uygulanmas için güçlü kan tlar mevcuttur. Bu a amada rekürrens en iyi ekilde anti-platelet ve anti-koagülan kombinasyonu ile önlenebilir (103).