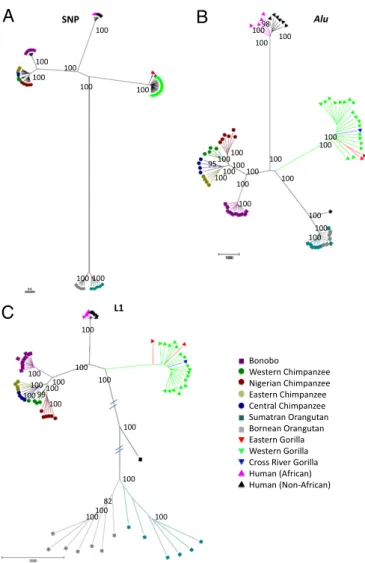
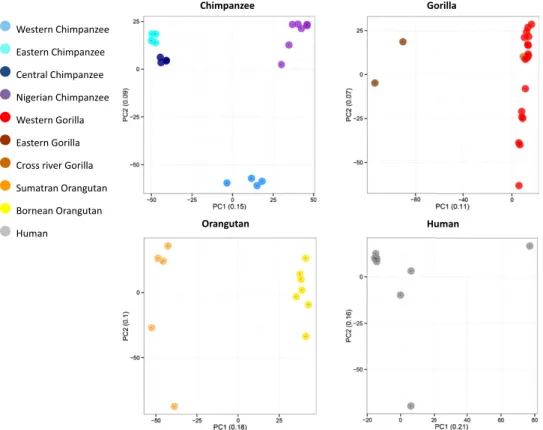
Rates and patterns of great ape retrotransposition
Tam metin
Şekil

Benzer Belgeler
Nejad gibi, Cezmi gibi, Vedad gibi, üçü de gençliklerinin tam orta sında, neslin o koldan devamını ümid ettiren birer mahsul bile veremeden sönüp giden
Pif, Pierfrancesco Diliberto, returns behind the camera after “La mafia uccide solo d’estate” / “The Mafia kills only in summer”, to direct “In guerra per amore” / “In
Bifosfonat ilaçlar intravenöz uygulamayla kanser tedavilerinde, oral alımla da osteopöröz tedavilerinde kullanılırlar.. Amino yan zinciri olan ve olmayan bifosfonatlar
Varolan spesifik periodontal durumun dikkatlice analizinden sonra hekim hasta için gerekli tüm gerekli işlemleri içeren bir tedavi planı ve faz 1 tedavi için gerekli
Kafkas Dağları da Avrupa ve Asya ayrı, ve en adaşı olan Kafkas ırkı Karpat Dağları , Orta ve Güney Avrupa'da büyük bir dağ. Alps Orta
Master of Science (MSc): 1 (Near East University, Institute of Health Sciences) Doctorate Thesis (Ph.D.): 8 (Hacettepe University, Institute of Health Sciences)1. Co-Referee in
• Suppose we think we can sell 50,000 cans per year at a price of $4 per can. It costs us about $2.5 per can, and such type of product has only a three-year life. We require a
Då landstingsstyrelsen inte avger några bedömningar avseende möjligheterna att uppnå fullmäktiges mål för verksamheten saknar vi underlag att uttala oss om utsikterna om
