T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
TIBBİ GENETİK ANABİLİM DALI
Kalıtsal Trombositopeni Hastalarının Hedeflenmiş Yeni Nesil Dizi
Analizi Sonuçlarının Retrospektif Olarak Değerlendirilmesi
SEMİH AŞIKOVALI
DANIŞMAN
DOÇ. DR. EMİN KARACA
İZMİR 2020
ÖNSÖZ
Tez çalışmamın gerçekleşmesi sürecinde bana yol gösteren, zamanını, tecrübesini, yardımını hiçbir zaman eksik etmeyen, birlikte çalışma fırsatı bulduğum için kendimi şanslı hissettiğim, anlayışlı, sabırlı ve hoşgörülü tutumuyla her zaman yanımda olan danışman hocam Doç. Dr. Emin KARACA’ya
Uzmanlık eğitim sürecinde, bilgi ve deneyimleriyle bana yol gösteren, kendimi geliştirmem için yardımlarını çaba harcayan, sadece bugüne değil geleceğe de hazır olmamı sağlayan başta Tıbbi Genetik Anabilim Dalı Başkanı Prof. Dr. Haluk Akın olmak üzere; Prof. Dr. Cihangir Özkınay’a, Prof. Dr. Ferda Özkınay’a, Prof. Dr. Özgür Çoğulu’ya, Prof. Dr. Hüseyin Onay’a, Doç. Dr. Burak Durmaz’a, Doç. Dr. Asude Durmaz’a, Doç. Dr. Ayça Aykut’a
Tez çalışmam sırasında desteklerini ve tecrübelerini benimle paylaşan Prof. Dr. Deniz Yılmaz Karapınar’a, Prof. Dr. Ayşegül Ünüvar’a, Prof. Dr. Tiraje Celkan’a
Eğitimim süresince bilgi ve tecrübelerini benimle paylaşan, çocuk hastalara yaklaşım konusunda kendimi geliştirmeme fırsat tanıyan Doç. Dr. Tahir Atik’e, Dr. Esra Işık’a, Dr. Bilçağ Akgün’e, Dr. Durdugül Ayyıldız’a, Dr. Enise Durmuşalioğlu’na, Dr. Melis Demir Köse’ye, Dr. Semih Özgüç Şimşir’e, Dr. Ayhan Keçelioğlu’na
Uzmanlık eğitimim boyunca bitmek bilmeyen sorularıma sabırla verdikleri cevaplar ve paylaştıkları deneyimleri için Uzm. Dr. Erhan Parıltay ve Uzm. Dr. Aslı Ece Solmaz’a
Asistanlık eğitimi süresince birlikte çalışmaktan mutluluk duyduğum, Dr. Hasan Taşlıdere’ye, Dr. Ayşenur Kavasoğlu’na, Dr. Hilmi Bolat’a, Dr. Tuba Türk’e Dr. Emine İpek Ceylan’a, Dr. Elif Uzay’a, Dr. Zehra Cengisiz’e, Dr. Yasemin Karaca’ya, Dr. Gizem Kök’e, Dr. Duygu Arıcan’a, Dr. Anıl Kalyoncu’ya, Dr. Mert Pekerbaş’a, Dr. Burak Aşçıoğlu’na
Eğitim sürecim boyunca bana işin pratik noktalarını öğreten, yardımcı olan ve tecrübelerini aktaran Ege Üniversitesi Tıbbi Genetik ailesinde teknisyen, biyolog, sekreter ve personel olarak görev yapan tüm çalışma arkadaşlarıma,
Tüm hayatım boyunca beni destekleyen, her zaman yanımda olan tüm arkadaşlarıma, dualarını üzerimden eksik etmeyen sevgili anne ve babama, sadece üç harften oluşan abi sözcüğünün içine dünyaları sığdıran canım abime
iii İÇİNDEKİLER ÖNSÖZ ... ii İÇİNDEKİLER ... iii ÖZET ... vi ABSTRACT ... vii
TABLOLAR LİSTESİ ... viii
ŞEKİLLER LİSTESİ ... ix KISALTMALAR LİSTESİ ... xi 1. GİRİŞ ... 1 2. GENEL BİLGİLER ... 2 2.1 MEGAKARYOPOEZ VE TROMBOPOEZ ... 2 2.2 TROMBOSİTOPENİLERE GİRİŞ ... 5 2.3 KALITSAL TROMBOSİTOPENİLER ... 6
2.3.1 KALITSAL TROMBOSİTOPENİLERİN TANISINDA YENİ NESİL DİZİ ANALİZİNİN KULLANIMI ... 7
2.3.2 KALITSAL TROMBOSİTOPENİLERİN GENETİK NEDENLERİ ... 8
2.3.2.1 MEGAKARYOSİT FARKLILAŞMASINDAKİ DEFEKTLER.... 10
2.3.2.1.1 Konjenital Amegakaryositik Trombositopeni ... 10
2.3.2.1.2 Trombosit Absent Radii Sendromu ... 11
2.3.2.1.3 Radioulnar Sinositozis ile Birlikte Giden Amegakaryositik Trombositopeni 12 2.3.2.2 MEGAKARYOSİT MATURASYONUNDAKİ BOZUKLUKLAR 13 2.3.2.2.1 Ailesel trombosit bozukluğu ve myeloid lösemilere yatkınlık . 13 2.3.2.2.2 ANKRD26 ilişkili Trombositopeni ... 14
2.3.2.2.3 Paris-Tousseau Trombositopenisi ve Jacobsen Sendromu ... 15
iv
2.3.2.2.5 GATA1 ilişkili hastalıklar ... 17
2.3.2.2.6 GFI1B ilişkili trombositopeni ... 19
2.3.2.2.7 Gri Trombosit Sendromu ... 19
2.3.2.2.8 SLFN14 ilişkili trombositopeni ... 20
2.3.2.2.9 FYB1 ilişkili trombositopeni ... 21
2.3.2.2.10 SRC ilişkili trombositopeni ... 21
2.3.2.3 TROMBOSİT SALINIMINDAKİ BOZUKLUKLAR ... 22
2.3.2.3.1 MYH9 ilişkili trombositopeni ... 22
2.3.2.3.2 ACTN1 ilişkili trombositopeni ... 24
2.3.2.3.3 FLNA ilişkili trombositopeni ... 24
2.3.2.3.4 Bernard-Soulier Sendromu ... 25
2.3.2.3.5 ITGA2B/ITGB3 ilişkili trombositopeni ... 26
2.3.2.3.6 TUBB1 ilişkili trombositopeni ... 27
2.3.2.3.7 TRPM7 ilişkili trombositopeni ... 27
2.3.2.3.8 TPM4 ilişkili trombositopeni ... 28
2.3.2.3.9 CYCS ilişkili trombositopeni ... 28
2.3.2.3.10 DIAPH1 ilişkili trombositopeni ... 28
2.3.2.3.11 PRKACG ilişkili trombositopeni ... 29
2.3.2.4 Azalmış trombosit ömrü ile ilişkili trombositopeniler ... 29
2.3.2.4.1 Tip 2B von Willebrand hastalığı ... 29
2.3.2.4.2 Wiskott- Aldrich Sendromu ... 29
2.3.2.5 Sebebi bilinmeyen defektler ... 31
2.3.2.5.1 Stormorken Sendromu ... 31
3. YÖNTEM ... 33
3.1 HASTA SEÇİMİ ... 33
3.2 MOLEKÜLER GENETİK ÇALIŞMALAR ... 33
3.3 VARYANT ÖNCELİKLENDİRME ... 35
v 4. BULGULAR ... 39 5. TARTIŞMA ... 62 6. SONUÇ VE ÖNERİLER ... 77 7. KAYNAKLAR ... 78 8. EKLER ... 95
vi ÖZET
Kalıtsal trombositopeniler, trombosit sayısının düşük olması ile karakterize, kanama eğilimi oluşturan heterojen hastalık grubudur. Bu hastalık grubunda 2010 yılına kadar 36 farklı form tanımlanmıştır. Genetik teknolojilerdeki gelişmeyle birlikte günümüzde en az 51 farklı hastalığın bu tabloya sebep olduğu bilinmektedir. Her bir hastalığın tedavisi ve prognozu birbirinden farklıdır.
Trombositopeni ile takip edilen olgularda önemli sorunlardan bir tanesi yanlış tanı ile takip edilme oranının yüksek olmasıdır. Yanlış tanı beraberinde yanlış medikal tedavi ve cerrahi girişimleri getirmektedir. Genetik testler olguları doğru tanıya bir adım daha yaklaştırmaktadır. Bu çalışmada, trombositopeni ön tanısı ile tarafımıza yönlendirilen 36 olgunun hedefe yönelik yeni nesil dizi analizi sonuçlarının retrospektif olarak değerlendirilmesi amaçlanmıştır.
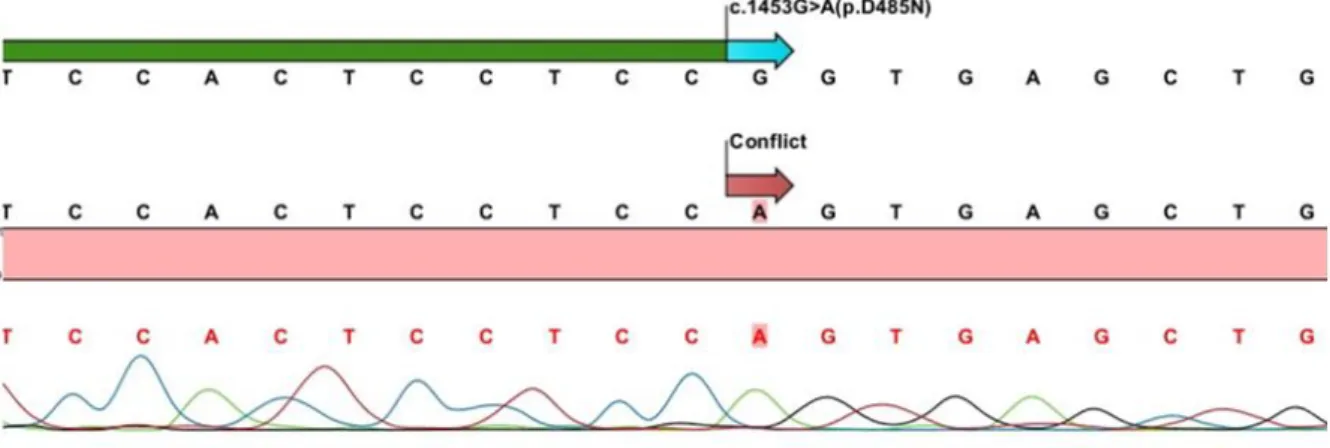
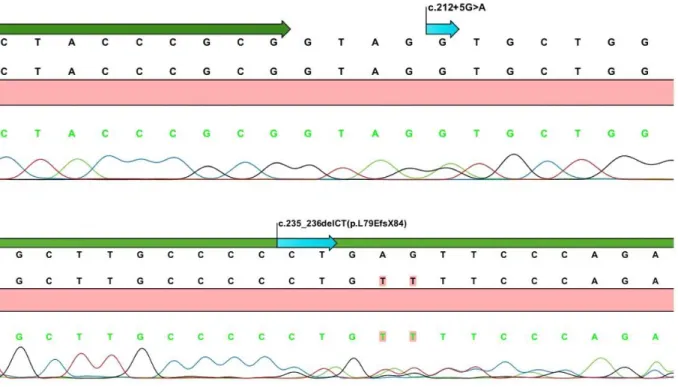
Çalışmaya alınan 36 olgunun 10’unda, kalıtsal trombositopeniye sebep olan genetik mutasyon mevcuttu. MYH9 geninde iki tanımlı (p.S96L, p.R702C), WAS geninde bir tanımlı (p.D485N), MPL geninde iki tanımlı (c.212+5G>A, p.L79EfsX84), GP1BA geninde bir tanımlı (p.N150S) ve NBEAL2 geninde iki yeni (c.7878+2G>A, p.G2520V) mutasyon saptanmıştır. Olguların klinik ve genetik özellikleri, genotip-fenotip korelasyonu açısından ele alındı.
Bu çalışma, kalıtsal trombositopeni hasta grubundaki mutasyon dağılımını inceleyen ülkemizdeki az sayıdaki çalışmadan biridir. Moleküler genetik çalışmaların olgulardaki klinik yaklaşıma etkisi değerlendirildi. Bu olgu grubunda moleküler tanı konulması, hastalara tedavi ve prognoz tahmini açısından önemli katkılar sunmakla birlikte preimplantasyon genetik tanı yoluyla sağlıkla kuşakların oluşmasına katkıda bulunacaktır.
Anahtar Kelimeler: Kalıtsal trombositopeni, yeni nesil dizi analizi, genotip-fenotip korelasyonu
vii ABSTRACT
Inherited thrombocytopenias are a heterogeneous group of diseases that are characterized by low platelet count which causes bleeding tendency. Until 2010, 36 different forms were defined in this disease group. Today, through to advances in genetic technologies, at least 51 different diseases are known to cause this condition. The treatment and prognosis of each disease are different.
As a result of the high rate of misdiagnosis, these cases undergo inappropriate treatment and surgical procedures. Genetic tests contribute to the correct diagnosis in these cases. In this study, we aimed to retrospectively evaluate the results of targeted next generation sequencing analysis of 36 patients who were referred with the initial diagnosis of thrombocytopenia retrospectively.
In the 10 of the 36 cases included in the study had a genetic mutation causing inherited thrombocytopenia. Mutations in the cases were as follows: Two previously reported (p.S96L, p.R702C) in MYH9 gene, one previously reported (p.D485N) in WAS gene, two previously reported (c.212+5G>A, p.L79EfsX84) in MPL gene, one previously reported (p.N150S) in GP1BA gene and two novel mutation (c.7878+2G>A, p.G2520V) in NBEAL2 gene. The clinical and genetic characteristics of the cases were discussed in terms of genotype phenotype correlation.
This study is one of the few studies in our country to showing distribution of mutations in inherited thrombocytopenia patient group. We evaluated the molecular genetic studies effect on the clinical approach these cases. Molecular genetic diagnosis provides important contributions for patients both in terms of treatment-prognosis prediction and growing of a healthy generation via preimplantation genetic diagnosis.
Keywords: Inherited thrombocytopenia, next-generation sequencing, genotype- phenotype correlation
viii TABLOLAR LİSTESİ
Tablo 1. Trombositopeniye neden olan ana mekanizmalar ... 6
Tablo 2. Kalıtsal Trombositopenilerin Patogenetik Mekanizmaları ... 9
Tablo 3. Konjenital amegakaryositik trombositopeni olgularında OMIM bulguları 10 Tablo 4. Trombosit absent radii sendromu olgularındaki OMIM bulguları ... 11
Tablo 5. RUSAT tip 1 olgularındaki OMIM bulguları... 12
Tablo 6. RUSAT tip 2 olgularındaki OMIM bulguları... 13
Tablo 7. FPD/AML olgularındaki OMIM bulguları... 14
Tablo 8. ANKRD26 ilişkili trombositopeni olgularındaki OMIM bulguları ... 14
Tablo 9. ANKRD26 genindeki 5’UTR bölgesinde tanımlı mutasyonlar ... 15
Tablo 10. Paris-Tousseau trombositopenisi olgularındaki OMIM bulguları... 16
Tablo 11. Jacobsen Sendromu olgularındaki OMIM bulguları ... 16
Tablo 12. ETV6 ilişkili trombositopeni olgularındaki OMIM bulguları ... 17
Tablo 13. XLTT olgularındaki OMIM bulguları ... 18
Tablo 14. XLANP olgularındaki OMIM bulguları... 18
Tablo 15. XLTDA olgularındaki OMIM bulguları ... 18
Tablo 16. GFI1B ilişkili trombositopeni olgularındaki OMIM bulguları ... 19
Tablo 17. Gri trombosit sendromu olgularındaki OMIM bulguları ... 20
Tablo 18. SLFN14 ilişkili trombositopeni olgularındaki OMIM bulguları ... 21
Tablo 19. FYB1 ilişkili trombositopeni olgularındaki OMIM bulguları ... 21
Tablo 20. SRC ilişkili trombositopeni olgularındaki OMIM bulguları ... 22
Tablo 21. MYH9 ilişkili trombositopeni olgularındaki OMIM bulguları ... 23
Tablo 22. Bernard-Soulier Sendromu olgularındaki OMIM bulguları ... 25
Tablo 23. Klasik Wiskott-Aldrich Sendromu olgularındaki OMIM bulguları ... 30
Tablo 24. X’e bağlı trombositopeni olgularındaki OMIM bulguları ... 31
Tablo 25. X’e bağlı nötropeni olgularındaki OMIM bulguları... 31
Tablo 26. Stormorken Sendromu olgularındaki OMIM bulguları... 32
Tablo 27. Hedef zenginleştirmede kullanılan panelin kapsadığı genler ... 34
Tablo 28. PCR koşulları ... 37
Tablo 29. Big-Dye Terminatör Kit PCR koşulları... 37
Tablo 30. Çalışmaya alınan 36 olgunun genel özellikleri ... 39
ix ŞEKİLLER LİSTESİ
Şekil 1. Hemapoetik kök hücrelerden kan hücreleri oluşumunda önerilen model ... 2
Şekil 2. Megakaryopoez ve trombopoezdeki ana moleküler mekanizmalar ... 4
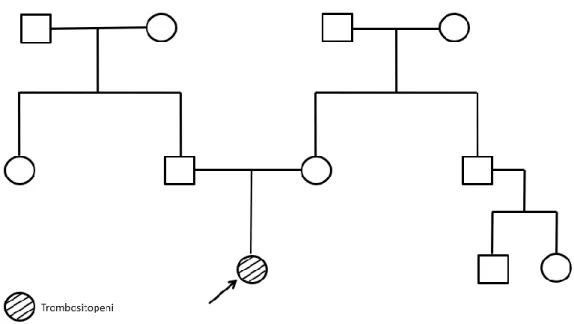
Şekil 3. Olgu 1'in aile ağacı ... 41
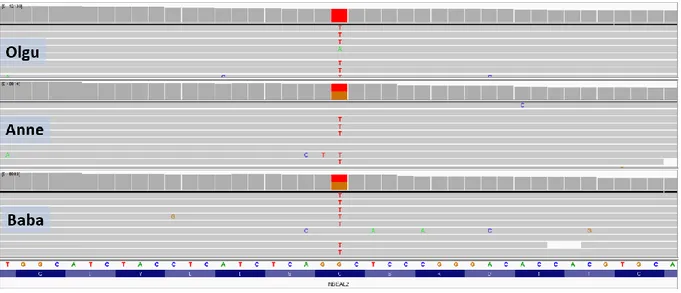
Şekil 4. Olgu 1'in Sanger dizi analizi görüntüsü ... 42
Şekil 5. Olgu 1'in segregasyon çalışması ... 42
Şekil 6. Olgu 3'ün aile ağacı ... 43
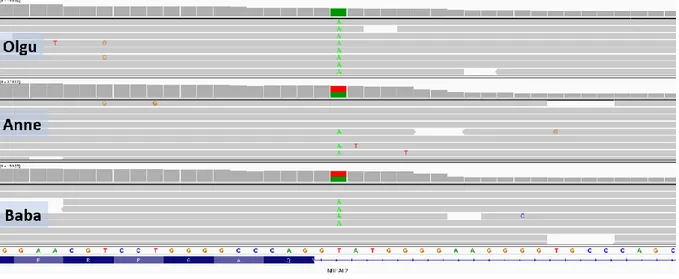
Şekil 7. Olgu 3'ün Sanger dizi analizi görüntüsü ... 44
Şekil 8. Olgu 3'ün segregasyon çalışması ... 44
Şekil 9. Olgu 5'in aile ağacı ... 45
Şekil 10. Olgu 5'in Sanger dizi analizi görüntüsü ... 46
Şekil 11. Olgu 5'in segregasyon çalışması ... 46
Şekil 12. Olgu 9'un aile ağacı ... 47
Şekil 13. Olgu 9'un Sanger dizi analizi görüntüsü ... 48
Şekil 14. Olgu 9'un segregasyon çalışması (I) ... 49
Şekil 15. Olgu 9'un segregasyon çalışması (II) ... 49
Şekil 16. Olgu 16'nın aile ağacı ... 50
Şekil 17. Olgu 16'nın Sanger dizi analizi görüntüsü ... 51
Şekil 18. Olgu 16'nın segregasyon çalışması ... 51
Şekil 19. Olgu 25'in aile ağacı ... 52
Şekil 20. Olgu 25'in Sanger dizi analizi görüntüsü ... 53
Şekil 21. Olgu 25'in segregasyon çalışması ... 53
Şekil 22. Olgu 28'in aile ağacı ... 54
Şekil 23. Olgu 28'in Sanger dizi analizi görüntüsü ... 55
Şekil 24. Olgu 28'in segregasyon çalışması ... 55
Şekil 25. Olgu 29'un aile ağacı ... 56
Şekil 26. Olgu 29'un Sanger dizi analizi görüntüsü ... 57
Şekil 27. Olgu 29'un YNDA görüntüsü ... 57
Şekil 28. Olgu 32'nin aile ağacı ... 58
Şekil 29. Olgu 32'nin Sanger dizi analizi görüntüsü ... 59
Şekil 30. Olgu 32'nin segregasyon çalışması ... 59
Şekil 31. Olgu 34'ün aile ağacı ... 60
x Şekil 33. Olgu 34'ün segregasyon çalışması ... 61
xi KISALTMALAR LİSTESİ
KT: Kalıtsal trombositopeni YNDA: Yeni nesil dizi analizi HKH: Hematopoetik kök hücre OLÖ: Ortak lenfoid öncül OMÖ: Ortak myeloid öncül
MEÖ: Megakaryosit/eritrosit öncül TPO: Trombopoetin
aa: Aminoasit
c-Mpl: onkogen myeloproliferatif lösemi virusu FOG1: Friend of GATA1
NFE2: Transkripsiyon faktörü p45 DMS: Demarkasyon membran sistemi ITP: İmmün trombositopenik purpura BSS: Bernard-Soulier Sendromu WHO: Dünya Sağlık Örgütü
ACMG: Amerikan Tıbbi Genetik ve Genomik Koleji KAMT: Konjenital amegakaryositik trombositopeni TAR: Trombosit absent radii
RUSAT: Radioulnar sinositoz ile birlikte giden amegakaryositik trombositopeni FPD/AML: Ailesel trombosit bozukluğu ve myeloid lösemilere yatkınlık ANKRD26: Ankyrin repeat domain 26 gene
PTT: Paris-Tousseau trombositopenisi JS: Jacobsen Sendromu
XLTT: X’ bağlı trombositopeni ve beta talasemi
XLANP: X’e bağlı anemiye eşlik eden-etmeyen nötropeni ve-veya trombosit bozuklukları
XLTDA: X’e bağlı trombositopeniye eşlik eden-etmeyen diseritropoetik anemi GPS: Gri trombosit sendromu
ADAP: Adezyon ve degranülasyon destekleyici adaptör proteini MYH9-RT: MYH9 ilişkili trombositopeni
FLNA: Filamin A
vWF: von Willebrand faktör Gp: Glikoprotein
xii mBSS: Monoallelik Bernard-Soulier Sendromu
vWD: von Willibrand Hastalığı
BDPLT16: Otozomal dominant trombosit tipi kanama bozukluğu 16 GT: Glanzmann thrombastenisi
stc: Sitokrom C
WAS: Wiskott-Aldrich Sendromu XLT: X’e bağlı trombositopeni XLN: X’e bağlı nötropeni
gnomAD: The Genome Aggregation Database ExAC: Exome aggregation Consortium PCR: Polimeraz zincir reaksiyonu NSVD: Normal spontan vaginal doğum
NMMHC-IIA: Kas dışı miyozin IIA ağır zincirini HD: N-terminal globuler head domain
ND: Neck domain TD: C-terminal domain
NHT: Helikal olmayan kuyruk
1 1. GİRİŞ
Kalıtsal trombositopeniler (KT), trombosit sayısının düşük olması ile karakterize, kanama eğilimi oluşturan heterojen hastalık grubudur. Bu hastalık grubuyla ilgili sahip olduğumuz bilgi birikimi son yıllarda önemli artış göstermiş, hastalıkların etyopatogenezlerinin daha iyi anlaşılmasını sağlamıştır. Başta kanama semptomları olmak üzere klinik değişkenliğin yüksek olması olguların tanı alma yaşını da etkilemektedir. Geniş olgu serilerinde ortalama tanı yaşları 21 yaş ve 37 yaş olarak belirtilmiştir (1). Kalıtsal trombositopenili hastaların kesin tanısının koyulması, tedaviye ve prognoza etkisi sebebiyle çok önemlidir. Örneğin; ANKRD26 ilişkili trombositopeni tanısı koyulan 78 olgunun incelendiği hasta serisinde, hastalardan 30’unun başlangıçta immun trombositopeni tanısı aldığı ve 16 olguya splenektomi de dahil olmak üzere gereksiz tedaviler verildiği belirtilmiştir (2). Konjenital amegakaryositik trombositopenide doğumda görülen ciddi trombositopeni hızlıca kemik iliği yetmezliğine ilerlediğinden kök hücre naklinin erken dönemde planlanması prognozu olumlu yönde etkileyecektir (3). Trombosit absent radii sendromunda ise doğumda şiddetli olan trombositopeni, yaş ilerledikçe düzeldiğinden sadece destekleyici tedaviler önerilmektedir (4). MYH9 gen mutasyonlarında trombopoetin-mimetik ilaçların kullanımı için klinik çalışmalar ise devam etmektedir (5). RUNX1, ANKRD26 ve ETV6 mutasyonlarında artmış myeloid lösemi riski açısından yakın takip gerekir. MYH9 gen mutasyonlarındaki işitme kaybı, nefropati gibi ekstra hematolojik bulguların erken tanınması komplikasyonların azaltılmasını sağlamaktadır (6). Bu sebeplerden dolayı KT hastalık grubunda doğru tanının erken dönemde konulması, dikkatli ve hedefe yönelik klinik takip önemlidir.
Kalıtsal trombositopeniler hakkında bilgi birikimimizi arttıran en önemli gelişme yeni nesil dizi analizinin (YNDA) rutin kullanıma girmiş olmasıdır. YNDA sayesinde, genomumuzdaki kodlayıcı tüm ekzonların nükleotit dizilerine ya da genomumuzdaki tüm nükleotit dizisine ulaşmak çok daha hızlı ve ucuz bir hale gelmiştir. Örneğin; GFI1b, STIM1,
FYB, SLFN14, ETV6, DIAPH1 ve SRC genlerinin KT’ye sebep oldukları yüksek çıktılı
YNDA uygulamaları sayesinde keşfedilmiştir (7).
Bu çalışmada KT ön tanısı ile tarafımıza yönlendirilen 36 olgunun hedefe yönelik YNDA sonuçları retrospektif olarak değerlendirilmiştir. Olguların mutasyon dağılımı, genotip-fenotip korelasyonları ve klinik öyküleri ile bu hastalıklara dikkat çekerek, literatüre katkı sağlamak amaçlanmıştır.
2 2. GENEL BİLGİLER
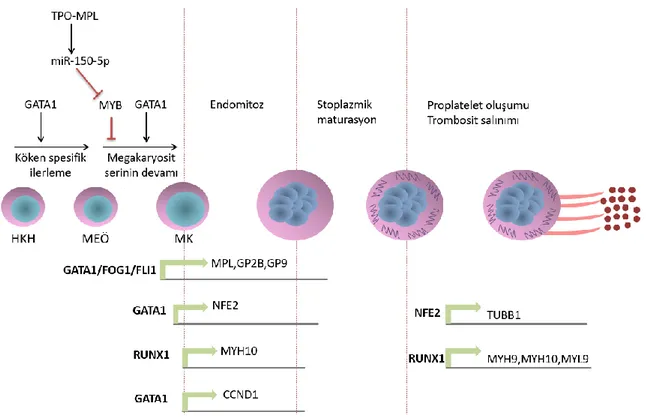
2.1 MEGAKARYOPOEZ VE TROMBOPOEZ
Megakaryopoez ve trombopoez, hematopoetik kök hücrelerden (HKH) proliferasyon, diferansiasyon ve matürasyon işlemlerinden sonra trombositlerin oluşum sürecidir. Bu 2 süreç hematopoezin önemli bir bölümünü oluşturur.
Şekil 1. Hemapoetik kök hücrelerden kan hücreleri oluşumunda önerilen model
Hematopoetik sistem, HKH’lerden kan hücrelerinin tamamının ömür boyu tedarikini sağlar. HKH’lerin çeşitli kan hücresi tiplerine farklılaşma süreci ağaç benzeri dallı bir yol haritası olarak kabul görmüştür (Şekil 1) (8). HKH’lerden megakaryosit oluşumuna giden süreçte, ilk dallanma noktası ortak lenfoid öncül (common lenfoid progenitor) (OLÖ) ve ortak myeloid öncül (common myeloid progenitor) (OMÖ) oluşumudur. Megakaryosit oluşumu sırasında myeloid seriye devam etmek, lenfoid seriden ayrılmak gereklidir. Bu ilk dallanma noktasında oluşan OMÖ hücreleri; megakaryosit/eritrosit öncülü (megakaryocyte/erythrocyte progenitor) (MEÖ) ve granülosit/makrofaj öncülü (granulocyte/macrophage progenitor) (GMÖ) hücreleri oluşturma yeteneğindedirler (8). İkinci dallanma noktasındaki MEÖ hücrelerinden megakaryositler ya da eritrositler oluşmaktadır.
3 Hematopoetik kök hücrelerden trombosit oluşumuna kadar geçen süreçte; transkripsiyon faktörleri, sitokinler, kemokinler ve hücre dışı matriks bileşenleri rol alırlar (9). Bu süreçte görevli ana moleküler mekanizmalar Şekil 2’de özetlenmiştir (9). Karaciğerde sentezlenen trombopoetin (TPO), megakaryopoez ve trombopoezi yönlendiren, megakaryosit çoğalması ve farklılaşmasını düzenleyen primer sitokindir (10). Yapılan kapsamlı çalışmalar; TPO’nun megakaryosit büyümesi, farklılaşması ve buna bağlı olarak periferik kan trombosit düzeylerinin korunması için vazgeçilmez olduğunu ortaya koymuşlardır (11,12). TPO, eritropoietin-benzer bölge (1.-153. rezidü) ve karbonhidrat zengin bölgeden (154.-332. rezidü) oluşan 4 heliksli yapıda sitokin süper ailesinin bir üyesidir (13). İlk olarak 353 aminoasit (aa) uzunluğunda bir öncül olarak sentezlenir. Sinyal peptidi kısmı olan 21 aa’lik kısmı ayrıldığında 332 aa içerir ve glikozilasyona uğrar (14). Bu glikoprotein depolanmadan doğrudan dolaşıma salınır. TPO reseptörü ise, onkogen myeloproliferatif lösemi virüsünün (c-Mpl) hücresel homologu olduğu için MPL olarak da bilinir, distal ve proksimal olarak 2 bölgeden oluşur (11). Normalde inaktif dimer şeklinde bulunan TPO reseptörünün distal sitokin reseptörüne ligand bağlandığında reseptör aktifleşir (15). Aktifleşen reseptör başta JAK-STAT yolağı olmak üzere birçok sinyal yolağını aktifleştirir ve HKH’lerden megakaryosit oluşumuna kadar geçen tüm süreci doza bağımlı olarak aktifleştirir (12).
Transkripsiyon faktörleri ise hematopoezin köken spesifik olarak ilerlemesinde önemli rol alırlar. Bunlar içinde GATA1, eritroid ve megakaryoid farklılaşmada en temel rolü oynar. Embriyonik kök hücreler kullanılarak yapılan çalışmalarda, GATA1’in çalışmadığı durumlarda MEÖ hücrelerinin olgunlaşmasında bozulmalar olduğu gösterilmiştir (16). FOG1 (Friend of GATA1) ise bu süreçlerde GATA1’in kofaktörü olarak görev alır (17). Diğer transkripsiyon faktörlerinin ise, MEÖ hücrelerinin dengeli bir biçimde üretilmesine katkıda bulunduğu söylenebilir. Bu dengenin korunmasında MYB protoonkogeni ana regülatör olarak görev yapar (9). MYB, eritroid serinin transkripsiyon faktörü olan KLF1’in ekspresyonunu arttırarak eritropoezi indükler (18). Ayrıca, megakaryopoezi destekleyen transkripsiyon faktörü MAF’ın ekspresyonunu azaltan miR-486-3p, MYB tarafından indüklenir (19). Toplam sonuca bakıldığında MYB, MEÖ hücrelerin megakaryosit seriye devam etmesini kısıtlar. MYB’nin fonksiyon kaybına uğratıldığı deneylerde, megakaryopoez ve trombopoezin arttığı gösterilmiştir (20). Buna karşılık TPO, miR150-5p üretimini arttırak MYB’yi baskılar ve megakaryopoezi destekler (21). Megakaryosit seri yönünde ilerleme olurken, MYB down regülasyonu FLI1 ekspresyonunu mümkün kılar. FLI1 ise MPL, ITGA2B, GP2B, GP9 gibi megakaryosit ve platelet spesifik reseptörlerin ekspresyonunu arttırmak için GATA1-FOG1 kompleksi ve ETS1 ile birlikte hareket eder (22,23).
4 Megakaryositlerin matürasyonu ve trombosit üretiminin kontrolünde transkripsiyon faktörlerinden RUNX1 ve GATA1 önemli bir yere sahiptirler. RUNX1; MYH9, MYL9 ve
MYH10 ekspresyonunu düzenleyerek megakaryosit olgunlaşması sırasında poliploidizasyonu
ve proplatelet formasyonunun oluşumunu etkiler (24). RUNX1 ayrıca KLF1’i epigenetik olarak baskılayarak OMÖ hücrelerinden eritrosit seriye gidişatı sınırlandırır (25). GATA1 ise siklin D1 kodlayan genin (CCND1) ekspresyonunu arttırarak poliploidizasyonu düzenler (26). GATA1 ayrıca transkripsiyon faktörü p45 (NFE2)’in ekspresyonunu da aktifleştirir (9). NFE2, proplatelet oluşumu ve trombosit üretimi için gerekli birkaç genin ekspresyonunu (mikrotübüllerin ana yapısını oluşturan TUBB1 gibi) düzenler (27).
Şekil 2. Megakaryopoez ve trombopoezdeki ana moleküler mekanizmalar
(HKH: Hematopoetik kök hücre, MEÖ: Myeloid/eritrosit öncülü, MK: Megakaryosit)
Endomitoz ve proplatelet oluşumu, trombosit üretiminde megakaryosit öncül hücrelerinin geçirmesi gereken 2 önemli adımdır. Endomitoz, karyokinez ve sitokinez evrelerinin görülmediği, G1-S-G2-M evrelerini takiben geç anafazda duraksayan hücrelerin tekrar G1 evresine döndüğü mitozun özgün bir formudur (28). Hücrenin toplam DNA içeriği 2N-4N-8N-16N-32N-64N olarak birikir (29). Böylelikle hücre bölünmesi olmaksızın, tekrarlayan DNA replikasyonları ile hücre poliploid hale gelir. Nükleer poliploidizasyondan sonra megakaryositler, proplatelet adı verilen stoplazmik uzantılar oluştururlar. Bu uzantılar başlangıçta kalın olmalarına rağmen sonrasında incelmek için uzayıp kompleks dallanma
5 yapıları oluştururlar, trombositlerin sinüzoidal kan damarlarına salınmasına izin veren eşsiz bir mimariye sahiptirler (30). Trombositlerin dolaşıma geçme süresinde demarkasyon membran sistemi (DMS), mikrotübüller ve aktin filamanlar önemli rol oynarlar. DMS, trombosit oluşumu için havuz görevi üstlenen kıvrıntılı tübüllerden oluşan membran kanalları ağıdır. Mikrotübüller mitokondri, granül ve diğer veziküler organellerin trombositlere taşınmasını sağlarlar (31). Aktin filamanlar ise proplatelet dallanması için gereklidirler (30). Tüm olgunlaşma süresi bittiğinde trombositler dolaşımdaki görevlerine başlarlar.
2.2 TROMBOSİTOPENİLERE GİRİŞ
Trombopoetin’in ana düzenleyeci olduğu süreçte, megakaryositler; kemik iliğindeki üretim ve olgunlaşma döneminden sonra trombositleri oluştururlar. Trombositler, sadece pıhtılaşma sürecinde görev alan bir tıkaç oluşturmakla kalmazlar, aynı zamanda inflamasyon, immunite ve kanser biyolojisinde de çok önemli görev üstlenirler (32). Trombosit sayısı, yaş, etnik köken, cinsiyet, genetik ve çevresel faktörlerden etkilendiği için kişiden kişiye farklılıklar gösterebilir (33). Sağlıklı kişilerde trombosit sayısının 150-450.000/mm3 olması
beklenir. Genel olarak 150.000/mm3 altındaki sayılar trombositopeni olarak kabul edilir.
Trombosit değerleri 100-150.000/mm3 arasında olduğunda genellikle klinik bulgu
gözlenmezken, 10.000/mm3 değerinin altına düştüğünde ise ciddi kafa içi kanamalar
gözlenebilir. Düşük trombosit sayısı; epistaksis, peteşi, mukokütanöz kanamalar, hematüri ve menoraji gibi farklı semptomlarla kendini gösterebilir. Trombositopeniye neden olan ana mekanizmalar Tablo 1’de özetlenmiştir (34).
6 Tablo 1. Trombositopeniye neden olan ana mekanizmalar
Yalancı Trombositopeni
EDTA'ya bağlı in vitro trombosit aglütinasyonu Dev trombositlerin otomatik cihazlarda sayılamaması Sekestrasyon
Splenomegali (Portal Hipertansiyon) Kemik İliği Yetersiz Üretimi
Enfeksiyonlar (Epstein-Barr virus, cytomegalovirus, hepatit C, parvovirus B19, helicobacter pylori, insan immun yetmezlik virusu, bruselloz, tüberküloz, tifo
İlaçlar ve Medikal Tedavi (Antibiyotikler, alkol, kemoterapi, radyasyon) Beslenme yetersizlikleri (Folat, vitamin B12 eksiklikleri)
Karaciğer Hastalıkları
Kemik İliği Yetersizliği Sendromları (Aplastik anemi, Fankoni anemisi, diskeratosis konjenita, Diamond-Blackfan anemisi, Shwacman-Diamond Sendromu
Hematolojik Hastalıklar (Lenfoma, lösemi, myelodisplastik sendrom) Kemik iliğinin tümör ile infiltre olması
Kalıtsal Trombositopeniler Trombosit Yıkımında Artış İmmün trombositopenik purpura
İlaca bağlı immun trombositopenik purpura (kinin, non-steroid yapıda antienflamatuar ilaçlar, glikoprotein IIb/IIIa inhibitörleri
Heparine bağlı immun trombositopenik purpura
Trombotik trombositopenik purpura/ Hemolitik üremik sendrom Atipik hemolitik üremik sendrom
İlaca bağlı trombotik trombositopenik purpura (mitomisin C, gemcitabin, oksaplatin) Yaygın intravasküler koagülasyon
Transfüzyon sonrası oluşan purpura
Otoimmun hastalık ilişkili trombositopeni (sistemik lupus eritamatozus, antifosfolipid antikor sendromu, tiroid hastalıkları, Evans Sendromu)
Mekanik yıkıma bağlı (kardiyopulmoner bypass, intraaortic balon pompası)
2.3 KALITSAL TROMBOSİTOPENİLER
Trombosit sayısının normal sınırlarda tutulması, megakaryopoez ve trombopoez yoluyla üretilen trombositler ile yaşlanan trombositlerin kandan uzaklaştırılması arasındaki hassas dengenin korunmasına bağlıdır. KT bu uyumun genetik bir mutasyon tarafından bozulduğu durumlarda ortaya çıkar. Hastalar, doğumdan birkaç hafta sonra gelişen şiddetli kanamalar ile başvurabileceği gibi, ilerleyen yaşlarda rutin kan testi sırasında tesadüfen fark edilen düşük trombosit değerleri ile de başvurabilirler (35).
Trombosit sayısı düşük hastalara doğru tanının konulması tedavi seçiminde kritik bir öneme sahiptir. KT’ler en sık immun trombositopenik purpura (ITP) ile karıştırılırlar (35). ITP’de trombositlere karşı gelişen otoantikorların T lenfosit aracılı lizisi ve trombosit
7 fagositozu gözlenir (36). ITP hastalarının trombosit sayılarının 10.000/mm3 altına inip hayati tehlike oluşturmasını önlemek için intravenöz gamaglobulin, kortikosteroid gibi yoğun tedaviler alması gerekebilir (36). KT olgularına doğru ve erken tanı konulması hem gereksiz tedaviler verilmesini önler hem de hastaların klinik takibinin uygun yapılmasını sağlar.
Kalıtsal trombositopeniler, düşük trombosit sayısı ile karakterize, trombosit büyüklüğünün farklılık gösterebildiği, şiddeti değişken kanama semptomları görülen heterojen bir hastalık grubudur (37). Hastalığın nadir görüldüğü düşünülse de İtalyan populasyonununda yapılan bir çalışmada tahmini sıklığının en az 2.7/100.000 olduğu belirtilmiştir (38). Bu hastalık grubunda ilk olarak 1948 yılında Bernard-Soulier Sendromu (BSS) tanımlanmıştır (7). Günümüze kadar gelen süreçte teknolojik ilerlemeler sayesinde en az 51 geni etkileyen 33 farklı KT formu tanımlanmıştır (7). Bu formların her birinin ortaya çıkış sebebi, klinik seyri ve sekonder semptomları farklılıklar gösterebilir. Örneğin bu hastalık grubu içinde prevelansları sırasıyla %3, %18, %5 olarak tahmin edilen RUNX1, ANKRD26,
ETV6 gen mutasyonlarında artmış myeloid lösemi riski bulunmaktadır (7). Bu 3 genin
germline mutasyonları Dünya Sağlık Örgütünün (WHO) 2016 yılındaki “myeloid neoplazm ve akut lösemi” sınıflandırmasının revizyonunda “önceden var olan trombosit bozuklukları ve myeloid neoplazilere doğuştan yatkınlık” olarak yeni bir kategoride değerlendirilmiştir (39). Bu sebeple sporadik myeloid lösemi aile öyküsü olan olgular dikkatle değerlendirilmelidir. Bu olgular için kök hücre nakli tek küratif tedavi seçeneği olmakla beraber aile içerisinden yapılacak nakillerde genetik taşıyıcılık açısından dikkatli olmak gerekir.
2.3.1 KALITSAL TROMBOSİTOPENİLERİN TANISINDA YENİ NESİL DİZİ ANALİZİNİN KULLANIMI
Genetik teknolojilerin gelişimiyle birlikte KT’ler hakkındaki bilgi birikimi katlanarak artmıştır. Birinci nesil dizi analizi olan Sanger dizilemenin yüksek maliyet ve düşük çıktı dezavantajlarının, YNDA ile ortadan kaybolması, düşük maliyet ile yüksek çıktılı sonuçların alınması açısından çok önemlidir. YNDA ile genomun tamamına (tüm genom dizileme), genomdaki tüm protein kodlayan bölgelere (tüm ekzom dizileme) ya da bir hastalık-hastalık grubundan sorumlu tutulan tüm bölgelere özel (panel dizileme) çalışmalar planlanabilir. Özellikle son 10 yılda tüm ekzom dizileme ve tüm genom dizileme yöntemlerinin uygulanması, megakaryopoez ve trombopoez ile ilgili yeni genlerin keşfedilmesine ve etyopatogenezlerin aydınlatılmasına ışık tutmuştur (7). Ayrıca trombosit hastalıkları, kanama bozuklukları ya da hematolojik hastalıklar gibi daha geniş hedeflenmiş YNDA panel çalışmaları ile KT’lerin moleküler tanısı daha hızlı ve ucuz hale gelmiştir.
8 Test sayılarının ve elde edilen bilginin artması saptanan değişikliklerin klinik etkisini tahmin etme noktasında yeni zorlukları beraberinde getirmiştir. Bu aşamada genetik değişikliklerin tüm dünyada standart şekilde değerlendirilmesi ihtiyacı doğmuştur. Bu ihtiyaç neticesinde, Amerikan Tıbbi Genetik ve Genomik Koleji [American College of Medical Genetics and Genomics (ACMG)] 2015 yılında dizi analizi sonucunda saptanan değişikliklerin Mendeliyan hastalıklarda yorumlanmasında kullanılmak üzere bir kılavuz oluşturmuştur (40).
Dizi analizinde saptanan varyantların klinik etkisinin değerlendirilmesinde ACMG’nin 2015 yılında oluşturduğu kılavuz, bir çerçeve oluşturmaktadır. Bu kılavuza göre varyantlar; patojenik, olası patojenik, önemi bilinmeyen, olası benign ve benign olarak 5 sınıfa ayrılmıştır. Sınıflandırma; popülasyon verileri, bilgisayar tahmin programları, segregasyon verileri gibi 28 parametreden oluşan sonuçların birlikte değerlendirilmesi ile yapılmaktadır. ACMG kriterleri raporlama sürecine önemli katkılar sağlasada saptanan varyantın klinik etkiler açısından değerlendirilmesinde klinik hekim-tıbbi genetik hekimi arasındaki değerlendirmeler nihai karar açısından büyük önem taşımaktadır.
2.3.2 KALITSAL TROMBOSİTOPENİLERİN GENETİK NEDENLERİ
Kalıtsal trombositopenilerin patogenezine bakıldığında çoğunluğunun, trombosit oluşum sürecindeki 1 ve/veya daha fazla adımdaki bozukluklardan kaynaklandığını söyleyebiliriz. Daha nadir olarak da trombosit ömrünün kısalmasıyla açıklanabilir. KT’lere neden olan patogenetik mekanizmalar Tablo 2’de özetlenmiştir (7).
9 Tablo 2. Kalıtsal Trombositopenilerin Patogenetik Mekanizmaları
Sorumlu Gen OMIM numarası Kalıtım Şekli Megakaryosit farklılaşmasındaki defektler
Konjenital Amegakaryositik Trombositopeni MPL 604498 OR
Trombosit Absent Radii Sendromu RBM8A 274000 OR
Radioulnar Sinositozis ile birlikte giden Amegakaryositik Trombositopeni HOXA11 MECOM 605432 616738 OD OD Megakaryosit maturasyonundaki defektler
Ailesel trombosit bozukluğu ve myeloid lösemilere
yatkınlık RUNX1 601399 OD
Trombositopeni 2 ANKRD26 188000 OD
Paris-Tousseau Trombositopenisi ve
Jacobsen Sendromu FLI1
188025
147791 OD
Trombositopeni 5 ETV6 616216 OD
X’e bağlı trombositopeni ve beta talasemi GATA1 314050 XLR X’e bağlı anemiye eşlik eden-etmeyen nötropeni
ve-veya trombosit bozuklukları GATA1 300835 XLR
X’e bağlı trombositopeniye eşlik eden-etmeyen
diseritropoietik anemi GATA1 300367 XLR
Trombosit tipi kanama bozukluğu 17 GFI1B 187900 OD
Gri Trombosit Sendromu NBEAL2 139090 OR
Trombosit tipi kanama bozukluğu 20 SLFN14 616913 OD
Trombositopeni 3 FYB 273900 OR
Trombositopeni 6 SRC 616937 OD
Trombosit Salınımındaki Bozukluklar
Trombosit tipi kanama bozukluğu 6 MYH9 155100 OD
Trombosit tipi kanama bozukluğu 15 ACTN1 615193 OD
FLNA ilişkili trombositopeni FLNA - XLD
Bernard-Soulier Sendromu GP1BA GP1BB GP9 231200 OR / OD
Trombosit tipi kanama bozukluğu 16 ITGA2B
ITGB3 187800 OD
10
TRPM7 ilişkili trombositopeni TRPM7 - -
TPM4 ilişkili trombositopeni TPM4 - -
Trombositopeni 4 CYCS 612004 OD
DIAPH1 ilişkili trombositopeni DIAPH1 - OD
Trombosit tipi kanama bozukluğu 19 PRKACG 616176 OR
Azalmış Trombosit Ömrü ile İlişkili Trombositopeniler
Tip 2B von Willebrand hastalığı VWF 613554 OD
Trombositopeni 1 WAS 313900 XLR
Sınıflandırılamayanlar
Stormorken Sendromu STIM1
ORAI1 185070 OD
(OR: Otozomal Resesif, OD: Otozomal Dominant, XLR: X’e bağlı Resesif, XLD: X’e bağlı Dominant)
2.3.2.1 MEGAKARYOSİT FARKLILAŞMASINDAKİ DEFEKTLER
2.3.2.1.1 Konjenital Amegakaryositik Trombositopeni
Konjenital Amegakaryositik Trombositopeni (MIM 604498) (KAMT), infant dönemde fiziksel anomali görülmeden izole trombositopeni ve megakaryositopeni ile karakterize, MPL geninin biallelik fonksiyon kaybettirici mutasyonları sonucunda ortaya çıkan nadir bir hastalıktır (41). Genellikle 4 yaşına kadar pansitopeni ve kemik iliği yetmezliğine ilerler. Bu olgulardaki OMIM bulguları Tablo 3’de belirtilmiştir (42). Günümüzde küratif tek tedavi kemik iliği transplantasyonudur. MPL geni 1p34 kromozom bölgesinde lokalizedir, 12 kodlayıcı ekzon içerir ve 635 aminoasitlik bir protein kodlar. Bu gendeki mutasyonların %80’i ilk 5 ekzonda görülmekle beraber, özellikle 2. ve 3. ekzondaki mutasyonlar tüm mutasyonların %65’lik bir kısmını oluşturmaktadır (3). TPO’nun reseptörü olan MPL, HKH ve megakaryosit öncülü hücrelerin hücre yüzeyinde eksprese olur bu hücrelerin farklılaşma süreçlerini destekler (20).
Tablo 3. Konjenital amegakaryositik trombositopeni olgularında OMIM bulguları İskelet Bulguları
Normal radius Nörolojik Bulgular
Hipoplastik serebellar vermis Hematolojik Bulgular
Doğumda ciddi trombositopeni, çocukluk çağında pansitopeni, megakaryositopeni, yüksek serum trombopoetin düzeyleri
11 2.3.2.1.2 Trombosit Absent Radii Sendromu
Trombosit Absent Radii (TAR) Sendromu (MIM 274000) doğumda başlayan şiddetli trombositopeniye eşlik eden bilateral radius yokluğu ve başparmak anomalileri ile karakterize nadir bir hastalıktır. KAMT’dan farklı olarak trombositopeni genellikle iki yaşına kadar düzelir ve transplantasyon gereksinimi göstermez (4). TAR Sendromu olgularındaki OMIM bulguları Tablo 4’te verilmiştir (42). Hastalığın temelindeki genetik bozukluk, kromozom 1q21.1 bölgesindeki en az 200 kilobazlık delesyona eşlik eden RBM8A geninin aktif düzenleyici bölgesinde bulunan iki tane tek nükleotit polimorfizminden birinin birlikteliğidir (43). Bu iki polimorfizm 5’ UTR bölgesindeki rs139428292 G>A ve ilk introndaki rs201779890 G>C’dir. RBM8A geni, olgunlaşmamış mRNA yıkımında görevli ekzon bağlantı kompleksinin subuniti olan Y14 proteinini kodlar. TAR sendromu aynı zamanda ekzon bağlantı kompleksinin eksikliğine bağlı olduğu gösterilen ilk insan hastalığıdır (43).
Tablo 4. Trombosit absent radii sendromu olgularındaki OMIM bulguları Baş-Boyun Bulguları
Brakisefali, mikrognati, strabismus, pitozis, küçük-kalkık burun Kardiyovasküler Sistem Bulguları
Fallot tetralojisi, atrial septal defekt, ventriküler septal defekt, aort koarktasyonu Gastrointestinal Sistem Bulguları
Pankreatik kistler, meckel divertikülü Genitoüriner Sistem Bulguları
Böbreğin aksiyel malrotasyonu, at nalı böbrek İskelet Bulguları
Bilateral radius yokluğu, ulna hipoplazisi/yokluğu, anormal humerus, diz subluksasyonu, patella dislokasyonu, femoral torsiyon, tibial torsiyon, fibula yokluğu, karpal kemik hipoplazisi/füzyonu, hipoplastik falankslar
Nörolojik Sistem Bulguları
İntrakranial kanama, myelinizasyonda gecikme, serebullum hipoplazisi, kavum septum pellucidum, korpus kallosum yokluğu, nöbetler, motor gelişmede gecikme, öğrenme bozukluğu
Hematolojik Bulgular
Doğumda başlayan trombositopeni, magakaryosit yokluğu, lökomoid granülositozis, eozinofili, anemi, hipersellüler kemik iliği
İmmünolojik Bulgular
12 2.3.2.1.3 Radioulnar Sinositozis ile Birlikte Giden Amegakaryositik Trombositopeni
Radioulnar sinositozis ile birlikte giden amegakaryositik trombositopeni (RUSAT), pansitopeniye ilerleyen trombositopeni ve radius ile ulna kemiklerinin proksimal füzyonu sebebiyle önkolda pronasyon ve supinasyon hareketlerinin kısıtlandığı nadir bir hastalıktır (44). HOXA11 genindeki mutasyonlar RUSAT tip 1 (MIM 605432), MECOM genindeki mutasyonlar ise RUSAT tip 2’ye (MIM 616738) sebep olurlar. Bu iki hastalığın OMIM bulguları Tablo 5 ve 6’da verilmiştir (42). Her iki tip de otozomal dominant olarak kalıtılır.
HOXA11 geni 7p15.2 kromozom bölgesinde lokalizedir, 2 kodlayıcı ekzon içerir ve 313
aminoasitlik bir protein kodlar. HOXA11 proteini DNA bağlama etkisine sahiptir, HKH’lerde eksprese edilir (45). Etkinliği azalmış HOXA11 proteininin megakaryosit farklılışmasını bozduğu in vitro koşullarda gösterilmiştir (45). HOXA11 genindeki varyantların hangi mekanizma ile RUSAT kliniğine sebep olduğu halen bilinmemektedir. MECOM geni ise RUSAT kliniği gösteren fakat HOXA11 geninde varyant saptanmayan 3 olgunun ileri genetik incelemeleri ile bulunmuştur (44). MECOM geni, 3q26.2 kromozom bölgesinde lokalizedir, 15’i kodlayıcı olmak üzere 17 ekzon içerir ve 1116 aminoasitlik bir protein kodlar. Bu gen normal gelişim ve onkogeneziste önemli rolleri olan MDS1 ve EVI1 gibi iki önemli transkripsiyon faktörünü kodlar (46). Yapılan son çalışmalarda EVI1 proteininin farklı fonksiyonel bölgelerindeki mutasyonların radioulnar sinositozdan, iskelet anormalliği görülmeyen ciddi kemik yetmezliğine kadar uzanan farklı kliniklerle ilişkili olabileceği bildirilmiştir (46).
Tablo 5. RUSAT tip 1 olgularındaki OMIM bulguları Baş-Boyun Bulguları
Sensörinöral işitme kaybı İskelet Bulguları
Düz asetabulum, kalça çıkığı, proksimal radio-ulnar sinositozis, ön kolda pronasyon ve supinasyon kısıtlılığı, radial/ulnar bowing, el 5.parmakta klinodaktili, sindaktili
Hematolojik Bulgular
13 Tablo 6. RUSAT tip 2 olgularındaki OMIM bulguları
Baş-Boyun Bulguları
Sönsörinöral işitme kaybı (bazı hastalarda) İskelet Bulguları
Bilaretal radioulnar sinositoz, önkolda pronasyon ve supinasyon kısıtlılığı, el parmaklarında kemik defekti olmadan üst üste binme, bazı hastalarda el 5. parmakta klinodaktili/orta falanksda kemik defekti, el 4. parmakta brakimezofalaji
Cilt Bulguları
Konjenital sistemik peteşi, konjenital ekimoz (bazı hastalarda) Hematolojik Bulgular
Trombositopeni, kemik iliğinde megakaryosit yokluğu, bazı hastalarda konjenital şiddetli anemi ve nötropeni
2.3.2.2 MEGAKARYOSİT MATURASYONUNDAKİ BOZUKLUKLAR
2.3.2.2.1 Ailesel trombosit bozukluğu ve myeloid lösemilere yatkınlık
Ailesel trombosit bozukluğu ve myeloid lösemilere yatkınlık (FPD/AML) (MIM 601399), özellikle akut myeloid lösemi riskini arttıran, kalitatif ve kantitatif trombosit bozuklukları ile giden otozomal dominant kalıtılan bir hastalıktır (47). RUNX1 genindeki mutasyonlar nu tablodan sorumludur. FDP/AML olgularında klinik seyir oldukça değişkenlik gösterse de aynı aile içinde benzer semptomların görülmesi beklenir. Trombositopeni ve trombosit disfonksiyonuna sekonder gelişen kanama eğilimi hafif-orta düzeydedir. Prognozu esas etkileyen myeloid neoplazilerdir. Bu olgulardaki OMIM bulguları Tablo 7’de verilmiştir (42).
RUNX1 geni 21q22 kromozom bölgesinde lokalizedir, 6 ekzon içerir ve 453
aminoasit kodlar. Genel olarak gendeki büyük delesyonlar haployetersizlik ile yanlış anlamlı mutasyonlar ise dominant negatif etki ile hastalığa yol açarlar (48). Dominant negatif etkiye sahip mutasyonlarda myeloid lösemi riski daha yüksektir (47). Kodlayıcı bölgelere dizi analizinin yanında, delesyon ve duplikasyonlar için kopya sayısı analizleri şüpheli olgularda genetik çalışmalara mutlaka dahil edilmelidir. Yaşam boyu myeloid neoplazi riski %40 olarak bildirilmiştir (49).
14 Tablo 7. FPD/AML olgularındaki OMIM bulguları
Kanama Bulguları
Epistaksis, kolay morarma Hematolojik Bulgular
Doğumda trombositopeni, anormal trombosit aggregasyonu, uzamış kanama zamanı, normal trombosit hacmi ve morfolojisi
Kanserler
Akut monositik lösemi, myelodisplazi, lenfosarkoma, lenfositik lenfoma, akut myelositik lösemi, nöroblastoma
2.3.2.2.2 ANKRD26 ilişkili Trombositopeni
ANKRD26 (ankyrin repeat domain 26 gene) ilişkili trombositopeni (trombositopeni 2) (MIM 188000), ılımlı trombositopeniye eşlik eden sendromik özelliklerin olmadığı ve trombosit boyutunun normal aralıkta seyrettiği otozomal dominant kalıtılan genetik bir hastalıktır (50). Olguların çoğunda hafif kanama bulguları görülür, yaşamı tehdit edici ciddi kanama beklenmez. Trombosit alfa granüllerinde azalma görülürken, in vitro trombosit agregasyon testleri normal sınırlardadır (50). Bu olgulardaki OMIM bulguları Tablo 8’de verilmiştir (42). Akut lösemi açısından normal populasyona göre 30 kat artmış risk taşıdıkları belirtilse de bu risk RUNX1 ve ETV6 gen mutasyonlarına göre daha düşüktür (50). ANKRD26 geni 10p12 kromozom bölgesinde lokalizedir, 34 ekzon içerir ve 1726 aminoasit kodlar.
ANKRD26 geninin 5’UTR bölgesinde yer alan mutasyonlar hastalığa neden olurlar (2).
Bugüne kadar tanımlanan mutasyonlar Tablo 9’da gösterilmiştir (51,52). Fakat; c.-140C>G mutasyonu insan gen mutasyonu veritabanında [human gene mutation database (HGMD)] kayıtlı olmasına rağmen, allel frekansının yüksek olması nedeniyle klinik etki değerlendirilirken dikkatli olmak gerekmektedir (52).
Tablo 8. ANKRD26 ilişkili trombositopeni olgularındaki OMIM bulguları Kanama Bulguları
Kolay morarma Hematolojik Bulgular
Trombositopeni (ortalama 42.000 /mm3), normal trombosit hacmi, beyaz küre sayısında artış, plazma trombopoetin düzeyinde armış
15 Tablo 9. ANKRD26 genindeki 5’UTR bölgesinde tanımlı mutasyonlar
c.-116C>T c.-119C>A c.-127delAT
c.-116C>G c.-125T>G c.-128G>C
c.-118C>T c.-126T>G c.-128G>A
c.-118C>A c.-127A>T c.-134G>A
c.-118C>G c.-127A>G c.-140C>G
Megakaryopoezin son evrelerinde 2 transkripsiyon faktörü, RUNX1 ve FLI1,
ANKRD26 geninin 5’UTR bölgesine bağlanarak bu geni baskılarlar ve proplatelet oluşumu
tam zamanında sonlanır (53). Bu 2 transkripsiyon faktörü ANKRD26 uyarısını baskılayamadıklarında, yani ANKRD26 genindeki fonksiyon arttırıcı mutasyonlarda, TPO/MPL sinyalinin arttığı ve özellikle MAPK yolağı üzerinden proplatelet oluşumunda hasarlar meydana geldiği belirtilmiştir (53).
2.3.2.2.3 Paris-Tousseau Trombositopenisi ve Jacobsen Sendromu
Paris-Tousseau trombositopenisi (PTT) (MIM 188025) ve Jacobsen Sendromu (JS) (MIM 147791), 11q23 kromozom bölgesindeki delesyonlar sebebiyle ortaya çıkan, otozomal dominant kalıtıma sahip, yakın ilişki gösteren iki hastalıktır. PTT’de trombositopeninin ön planda olduğu, büyük alfa granüllerine ve anormal morfolojiye sahip trombositler görülür. JS’ de ise trombositopeninin yanına delesyonun büyüklüğüne bağlı olarak psikomotor gerilik, kalp problemleri ile trigonosefali, epikantus, telekantus, geniş burun kökü, kısa burun, antevert burun delikleri, retrognati, aşağı yerleşimli kulaklar gibi dismorfik bulgular eşlik eder (54). Bu 2 hastalıktaki OMIM bulguları Tablo 10 ve 11 de verilmiştir (42). PTT benzeri klinik bulgular gösteren ve 11q23 bölgesinde delesyon saptanmayan bir hastada FLI1 geninde homozigot yanlış anlamlı mutasyon saptandığında, bu 2 sendromdaki trombositopeninin sebebinin FLI1 gen kaybı olduğu düşünülmüştür (55). FLI1 gen mutasyonlarının, FLI1’in hedef genleri olan GP6,GP9 ve ITGA2B’nin ekspresyonlarını azaltarak trombosit maturasyonunda bozukluk oluşturduğu gösterilmiştir (55).
16 Tablo 10. Paris-Tousseau trombositopenisi olgularındaki OMIM bulguları
Baş-Boyun Bulguları
Trigonosefali, mikrognati, pitozis Gastrointestinal Bulgular Pilor stenozu İskelet Bulguları Klinodaktili Nörolojik Bulgular Zihinsel yetersizlik Hematolojik Bulgular
Trombositopeni, ılımlı kanama diyatezi, uzamış kanama zamanı, periferik yaymada trombositlerde büyük alfa granüller, kemik iliğinde megakaryosit artışı
Tablo 11. Jacobsen Sendromu olgularındaki OMIM bulguları İntrauterin büyüme kısıtlılığı
Baş-boyun bulguları
Trigonosefali, mikrosefali, makrosefali, düz oksipital kemik, mikrognati, düşük yerleşimli kulaklar, epikantal katlantı, hipertelorizm, pitozis, iris kolobomu, iriste renk değişikliği, strabismus, telekantus, mikroftalmi, mikrokornea, koryoretinal kolobom, maküler hipoplazi, nazolakrimal kanal tıkanıkları, anormal kirpikler-kaşlar, kısa burun, deprese burun köprüsü, büyük ağız, kısa boyun
Kardiyovasküler Bulgular
Ventriküler septal defekt, atrial septal defekt Göğüs Bulguları
Pektus ekskavatum, eksik kaburgalar Abdomen Bulguları
Halka şeklinde pankreas, pilor stenozu Genitoüriner Bulgular
Hipospadias, kriptoorşidizm (erkek), labial-klitoral hipoplazi (kadın) Nörolojik Bulgular
Hipotoni, spastisite, öğrenme bozukluğu, hidrosefali, holoprosensefali Hematolojik Bulgular
Trombositopeni
2.3.2.2.4 ETV6 ilişkili Trombositopeni
ETV6 ilişkili trombositopeni (trombositopeni 5) (MIM 616216), ETV6 genindeki germline mutasyonların sebep olduğu, ılımlı trombositopenin yanında artmış hematolojik ve solid malignite riski ile karakterize otozomal dominant kalıtılan bir hastalıktır (56). Trombositopeni erken çocukluk döneminde başlangıç gösterirken, yaşam boyu artmış
17 malignite riski bulunmaktadır. Bu olgulardaki OMIM bulguları Tablo 12’de verilmiştir (42).
ETV6, gen ekspresyonu düzenlemesinde rol alan ETS transkripsiyon faktörleri ailesinin bir
üyesidir ve 12p13 kromozom bölgesinde lokalizedir. Yapısal yeniden düzenlemeler (translokasyon-delesyon) varlığında, artmış hematolojik malignite riski uzun zamandır iyi bilinmektedir (57). Trombositopeni sebebi olduğu ise son yıllarda yapılan çalışmalar ile bulunmuştur. ETV6’nın megakaryosit gelişiminde proplatelet oluşumunda önemli rol aldığı in vitro çalışmalar ile gösterilmiştir (48). Nedeni açıklanamayan trombositopeni ve malignite öyküsü olan, aralarında akrabalık bulunmayan 3 aileye yapılan tüm ekzom dizi analizi sonucu, bu klinik tablodan ETV6 genindeki germline varyantların sorumlu olduğu bulunmuştur (58). Bugüne kadar 22 ailede tanımlanan ETV6 germline mutasyonlarından p.P214L mutasyonu 5 ailede tanımlanmıştır (48). Olguların %20’sinde B hücreli ALL, %30’unda ise diğer hematolojik maligniteler gözlenmiştir (48).
Tablo 12. ETV6 ilişkili trombositopeni olgularındaki OMIM bulguları Kanama Bulguları
Epiktaksis, kolay morarma, peteşi Hemotolojik Bulgular
Trombositopeni, bazı hastalarda anemi ve nötropeni Kanserler
Artmış hematolojik malignite riski
2.3.2.2.5 GATA1 ilişkili hastalıklar
GATA1 genindeki mutasyonlar; X’ bağlı trombositopeni ve beta talasemi (XLTT)
(MIM 314050), X’e bağlı anemiye eşlik eden-etmeyen nötropeni ve-veya trombosit bozuklukları (XLANP) (MIM 300835) ve X’e bağlı trombositopeniye eşlik eden-etmeyen diseritropoetik anemi (XLTDA) (MIM 300367) olmak üzere 3 farklı klinik tablodan sorumludurlar (59). Bu 3 farklı hastalığa sahip olgulardaki OMIM bulguları Tablo 13-14 ve 15’te verilmiştir (42). GATA1; eritrosit, megakaryosit, eozinofil, mast hücreleri gibi hematopoietik hücre serilerinin normal gelişiminin regulasyonunu sağlayan bir transkripsiyon faktörüdür. GATA1 geni; Xp12.23 kromozom bölgesinde lokalizedir, 5 tanesi kodlayıcı olmak üzere 6 ekzon içerir ve 413 aminoasitlik bir protein kodlar (60). GATA1 proteini, N-terminal transkripsiyon aktivasyon bölgesi ve iki çinko parmağa (zinc finger) sahiptir. C-terminal çinko parmak bölgesi çoğu gen için DNA bağlama aktivitesine sahipken, N-terminal çinko parmak bölgesi ise özellikle FOG1 ve TAL1 gibi önemli transkripsiyon faktörlerinin DNA ile etkileşime geçtiği palindromik dizileri barındırır (61).
18 Germline GATA1 mutasyonlarının fenotipi, genotipe bağlı olarak farklılıklar gösterebilir. Literatürdeki varyantlar incelendiğinde, p.V205M, p.G208R, p.D218Y varyantları ciddi diseritropoietik anemi ve makrotrombositopeni; p.G208S, p.D218G, p.D218N varyantları ılımlı disertropoetik özellikler ile birlikte makrotrombositopeni; p.R216Q varyantı makrotrombositopeni ile beraber beta talasemi ve p.Arg216Trp varyantı konjenital eritropoetik porfiri ile birlikte giden makrotrombositopeni ile ilişkilendirilmiştir (60). Ayrıca ekzon 2’deki c.322G>C, c.220G>C, c.220+1delG ve c.2C>G splice bölge mutasyonları, trombosit bozukluklarından çok ciddi anemi bulgularıyla ilişkilendirilmiştir (62). GATA1 fenotip-genotip ilişkilerini anlayabilmek için daha fazla çalışma gerekmektedir. Tablo 13. XLTT olgularındaki OMIM bulguları
Kanama Bulguları
Kolay morarma, peteşi, epistaksis Cilt Bulguları
Fotosensitif büllöz dermatit, hirşutizm Gastrointestinal Sistem Bulguları Splenomegali
Hematolojik Bulgular
Trombositopeni, uzamış kanama zamanı, retikülositoz, HbA-HbF yüksekliği, artmış trombosit hacmi, trombosit alfa granüllerinde azalma, trombosit fonksiyonlarında azalma, trombosit aggregasyon çalışmalarının normal sonuçlanması, hemolitik anemi
Tablo 14. XLANP olgularındaki OMIM bulguları Hematolojik Bulgular
Makrositik anemi, değişken derecede nötropeni, HbF yüksekliği, periferik yaymada anormal eritsositler, anizositoz, makrositoz, poikilositoz, eliptositoz, fragmente eritrositler, kemik iliğinde eritrosit hiposellülaritesi, bazı hastalarda granülosit hiposellülaritesi-mikromogakaryositler-bozulmuş trombosit agregasyonu-anormal trombosit morfolojisi
Tablo 15. XLTDA olgularındaki OMIM bulguları Kanama Bulguları
Epistaksis, kolay morarma, peteşi Hematolojik Bulgular
Trombositopeni, makrotrombosit, trombosit granüllerinde azalma, trombosit membran komplekslerinde anormallik, trombositlerde artmış düz endoplazmik retikulum, anormal trombosit matürasyonu, trombosit fonksiyonlarında bozulma, yükselmiş trombopoetin düzeyleri, bazı hastalarda diseritropoetik anemi ve anormal eritrosit morfolojisi
19 2.3.2.2.6 GFI1B ilişkili trombositopeni
GFI1B ilişkili trombositopeni (trombosit tipi kanama bozukluğu 17) (MIM 187900) yine son yıllarda tanımlanan, gri trombosit sendromuna benzeyen, anormal trombosit fonksiyonları nedeniyle kanama eğilimi oluşturan ve otozomal dominant kalıtılan nadir bir hastalıktır (63). Bu olgulardaki OMIM bulguları Tablo 16’da verilmiştir (42). Elektron mikroskobu ile incelendiğinde, trombositlerde azalmış ya da yok olmuş alfa granüller görülürken, kemik iliği biyopsisinde artmış sayıda anormal megakaryosit görülür. GFI1b geni 9q34 kromozom bölgesinde lokalizedir, 6’sı protein kodlayan 11 ekzon içerir. GFI1b hedef genlerin düzenleyici ve hızlandırıcı bölgelerinde histon modifikasyonları ile transkripsiyonel baskılayıcı olarak görev yapar. Hematapoez sırasında erken evrelerde öncül hücrelerinin farklılaşma özelliklerini korumalarına yardım eder (64).
Tablo 16. GFI1B ilişkili trombositopeni olgularındaki OMIM bulguları Kanama Bulguları
Epistaksis, ekimoz, peteşi, gastrointestinal kanama Hematolojik Bulgular
Trombositopeni, artmış trombosit hacmi, gri trombositler, trombositlerde alfa granül azlığı-yokluğu, değişken derecelerde trombosit aggregasyonunda azalmalar, displastik özellikte megakaryositler, myelofibrozis, emperipolezis, trombosit faktör 4'te azalma, beta-tromboglobulinde azalma, trombositler CD34 ve CD42b ekspresyonunda artma
2.3.2.2.7 Gri Trombosit Sendromu
Gri trombosit sendromu (GPS) (MIM 139090), ılımlı kanama eğilimi ve trombositopeninin görüldüğü, alfa granül proteinlerindeki belirgin azalmadan dolayı trombositlerin mikroskopta gri renkli (soluk) izlendiği, NBEAL2 genindeki mutasyonların sebep olduğu otozomal resesif kalıtılan hastalıktır (65). Bu olgulardaki OMIM bulguları Tablo 17’de verilmiştir (42). NBEAL2 geni 3p21 kromozom bölgesinde bulunur, 54 ekzondan oluşur. GPS hastalarının kanama şiddetinde ve trombosit fonksiyon testlerine yanıtlarında hafif ile orta derecede değişkenlik göstermek üzere heterojenite bulunmaktadır (66). Genel olarak, protein üretimi durduran mutasyonların homozigot durumda bulunması, proteinde aminoasit değişikliği yapan birleşik heterozigot mutasyonlara göre daha ağır klinik bulgular ile birliktelik gösterir (67). Bununla beraber aynı ailenin farklı üyelerinde farklı şiddetle klinik
20 bulgular olması, şu an için bilmediğimiz diğer faktörlerinde fenotipi etkilediğini göstermektedir (68).
Tablo 17. Gri trombosit sendromu olgularındaki OMIM bulguları Kanama Bulguları
Epistaksis, kolay morarma, menoraji Abdomen Bulguları
Splenomegali Hematolojik Bulgular
Trombositopeni, agranüler trombositler, Wright-Giemsa boyamada gri renkte trombositler, kemik iliğinde normal sellülarite ve retiküler fibrozis, myelofibrozis
Laboratuvar Anormallikleri
Ortalama trombosit hacmi 13 fL, uzamış kanama zamanı, araşidonik asit ile normal trombosit aggregasyonu, kollagen ve trombin ile azalmış trombosit aggregasyonu, azalmış von willebrand faktör-trombosit faktör 4-trombospondin-fibrinojen-fibronektin, artmış serum vitamin B12 düzeyleri
2.3.2.2.8 SLFN14 ilişkili trombositopeni
SLFN14 ilişkili trombositopeni (trombosit tip kanama bozukluğu 20) (MIM 616913), yakın zamanda üç farklı aileden 12 kişide tanımlanan, makrotrombositopeni ve artmış kanama eğilimi oluşturan, otozomal dominant kalıtılan çok nadir bir hastalıktır (69). Bu olgulardaki OMIM bulguları Tablo 18’de verilmiştir (42). SLFN14 geni, kromozom 17q12 bölgesinde lokalizedir, 4’ü kodlayıcı 6 ekzon içerir, 912 aminoasit kodlar. Bugüne kadar homozigot hasta tanımlanmamasının, mutant allelin normal allel üzerindeki dominant negatif etkisiyle açıklanabileceği bildirilmiştir (70). Heterozigot mutasyon bulunan durumlarda protein ekspresyonun %65-80 oranında azaldığı ve proplatelet uzantılar ile magakaryosit olgunlaşmasını bozduğu gösterilmiştir (69,71). SLFN14’ün endoribonükloetik yıkım aktivitesinin, trombosit olgunlaşması sırasında ribozomal yıkıma katkıda bulunduğu düşünülmektedir (70).
Bugüne kadar tanımlanan 4 mutasyonun tamamı (p.Lys218Glu, p.Lys219Asn, p.Val220Asp, p.Arg223Trp) DNA-RNA metabolizması sırasında ATP-GTP bağlanmasını sağlayan “AAA bölgesinde” yer almaktadır. Bu bölgenin trombositopeni kliniği ile ilişkisi araştırılmaya devam etmektedir (72).
21 Tablo 18. SLFN14 ilişkili trombositopeni olgularındaki OMIM bulguları
Kanama Bulguları
Epistaksis, diş eti kanamaları, menoraji , kolay morarma Hematolojik bulgular
Artmış kanama eğilimi, trombositopeni, ADP-kollajen-PAR1 ile agregasyon cevabının azalması, trombosit dense granüllerinde azalma, ATP sekresyonununda azalma, hasarlı proplatelet formasyonu, megakaryosit matürasyonunda bozulma
2.3.2.2.9 FYB1 ilişkili trombositopeni
FYB1 ilişkili trombositopeni (trombositopeni 3) (MIM 273900), infant dönemde başlayan mikrotrombositopeni ile karakterize, aralarında yakın akrabalık bulunan Iraklı bir ailede yakın zamanda tanımlanmış, otozomal resesif kalıtılan bir hastalıktır (73). Bu olgulardaki OMIM bulguları Tablo 19’da verilmiştir (42). FYB1 geni trombositlerden eksprese edilen, adezyon ve degranülasyon destekleyici adaptör proteini (ADAP) kodlar (74). ADAP proteini, integrin AIIbB3’ ün iç-dış regulasyonunda ve integrin A2B1’in kollagen
kaynaklı aktivasyonunda rol oynamaktadır (74).
Tablo 19. FYB1 ilişkili trombositopeni olgularındaki OMIM bulguları Kanama Bulguları
Epistaksis, menoraji, peteşi Hematolojik Bulgular
Trombositopeni, küçük trombositler, megakaryosit maturasyon defekti, ılımlı artmış kanama eğilimi
2.3.2.2.10 SRC ilişkili trombositopeni
SRC ilişkili trombositopeni (trombositopeni 6) (MIM 616937), myelofibroz, kanama, kemik patolojileri ve trombositopenisi bulunan 9 hastada yakın zamanda tanımlanan otozomal dominant kalıtılan nadir bir hastalıktır (75). Bu olgulardaki OMIM bulguları Tablo 20’de verilmiştir (42). SRC geni 20q11.2 kromozom bölgesinde lokalizedir ve reseptör olmayan tirozin kinaz yapısında protoonkogen kodlar. SRC geninin somatik fonksiyon kazandırıcı mutasyonları başta kolon kanseri olmak üzere çeşitli insan kanser hücrelerinde görülebilir (76). Trombositopeni kliniği ise gendeki fonksiyon kazandırıcı yanlış anlamlı germline mutasyonlar sebebiyle oluşur (75). Yapılan hücre kültürü çalışmalarında, SRC genindeki p.E527K değişikliğinin self-inhibisyonu azaltıp, hücrenin aktin iskeletinde hasara yol açtığı
22 böylelikle olgunlaşmamış megakaryositlerdeki proplatelet oluşumunu bozduğu gösterilmiştir (75).
Tablo 20. SRC ilişkili trombositopeni olgularındaki OMIM bulguları Baş-Boyun Bulguları
Geniş alın, hipotolerizm, derin yerleşimli gözler, geniş aralıklı burun delikleri, diş gelişiminin olmaması
İskelet Bulguları Osteoporoz
Hematolojik Bulgular
Trombositopeni, genişlemiş trombositler, kusurlu megakaryopoez, bazı hastalarda myelofibrozis-trombosit alfa granüllerinde eksiklik
2.3.2.3 TROMBOSİT SALINIMINDAKİ BOZUKLUKLAR
2.3.2.3.1 MYH9 ilişkili trombositopeni
MYH9 ilişkili trombositopeni (MIM 155100) (MYH9-RT) makrotrombositopeni ve granülosit inklüzyonlarına, nefrit ya da sensörinöral işitme kaybının eşlik ettiği ya da etmediği otozomal dominant kalıtılan bir hastalıktır. Daha önceden farklı fenotipik özellikler gösterdikleri için her biri ayrı bir klinik tablo olarak ele alınan; May-Hegglin Sendromu (MIM 155100), Sebastian Sendromu (MIM 605249), Fechtner Sendromu (MIM 153640) ve Epstein Sendromu (MIM 153640) günümüzde MYH9-RT olarak tek bir başlık altında değerlendirilmektedirler (77). Bu olgulardaki OMIM bulguları Tablo 21’de verilmiştir (42).
MYH9 geni, 22q13 kromozom bölgesinde lokalize 41 ekzondan oluşur, kodladığı 1960
aminoasit ile kas içermeyen miyozin IIA’ nın ağır zincirini oluşturur (78). Miyozinler, özellikle de tip IIA ve IIB, kasılma kuvveti oluşturan aktin filamanlar ile etkileşime girerek proplatelet oluşumunda rol oynarlar (79). Bu gendeki mutasyonlar, proplatelet formasyonunun hasarıyla birlikte, trombositlerin kemik iliğinden erken ayrıldığı bir makrotrombositopeni tablosuna sebep olurlar (7).
23 Tablo 21. MYH9 ilişkili trombositopeni olgularındaki OMIM bulguları
Kanama Bulguları
Epistaksis, kolay morarma, menoraji Baş-Boyun bulguları
İşitme kaybı (bazı hastalarda) Kardiyovasküler Sistem
Miyokardial enfarktüs (koroner arter trombozuna sekonder) Hematolojik Bulgular
Ilımlı trombositopeni, uzamış kanama zamanı, ortalama trombosit hacmi 12.5 fL, epinefrin-ADP-kollajen-ristosetin ile normal trombosit agregasyonu
MYH9 ilişkili trombositopeni tablosundan etkilenen tüm olgularda konjenital trombositopeni, trombositlerde makrositoz ve granülosit stoplazmalarında inklüzyon cisimleri görülür. Olguların az bir kısmında bu hematolojik değişiklikler yaşam boyunca hastalığın tek belirtisi olarak kalırken, çok büyük kısmında sensörinöral sağırlık, böbrek hastalıkları, presenil katarakt ve karaciğer enzim yüksekliği gibi geç başlangıçlı belirtiler gözlenir (6). İşitme kaybı, geç başlangıçlı bulgulardan en sık görülenidir ve ilerleyici seyreder (80). Ortalama 33 yaşında vakaların %50’sinde, zaman içinde ise tüm hastalarda duyma kaybı gelişmesi beklenir (6). Olguların yaklaşık %25’inde birkaç yıl içinde son dönem böbrek yetmezliğine giden proteinürik nefropati gözlenir (6). Olguların %20’sinde gözlenen presenil katarakt %75 oranında bilateraldir (6). Olguların yaklaşık yarısında gözlenen transaminaz ve gama glumatil transferaz yüksekliği genellikle karaciğer yetmezliğine ilerlemez (81).
Makrotrombositopeninin yanında hematolojik olmayan klinik bulgular varlığında MYH9-RT tanısı koymak nispeten kolaydır. Sporadik olguların yüksek sıklıkta olması (%35), düşük trombosit sayısının erişkin dönemde fark edilmesi ve otomatik kan sayım cihazlarının makrositozu atlaması tanıyı zorlaştırabilir (82). Bu olgular immun trombositopeniler ile zaman zaman karıştırılabilirler. Bu durum immunsupresif ilaç kullanımı ve splenektomi gibi tedavilerin gereksiz uygulanmasına yol açar (6).
Yakın zamanda yapılan MYH9 geninde mutasyona sahip 255 olgunun yer aldığı geniş çaplı bir çalışmada, olguların %85’inden 7 tane MYH9 genotipinin sorumlu olduğu gösterilmiştir (6). Genin 702. aminoasiti olan argininin (p.R702) yanlış anlamlı mutasyonları, 4. dekattan önce son dönem böbrek yetmezliği ve ciddi işitme kaybının görüldüğü ağır hastalık fenotipi ile ilişkilendirilmiştir (6). Geç başlangıçlı belirtilerin görülme riskinin yüksek olduğu p.D1424H ve 1165. Aminoasit olan argininin yanlış anlamlı (p.R1165) mutasyonlarında, işitme kaybı açısından yüksek risk bulunurken, böbrek hasarı ve katarakt riski daha düşüktür (6). Çerçeve kaybı mutasyonları (frameshift) ve protein üretimi durduran