INDEEDopt: a deep learning-based ReaxFF parameterization framework
Tam metin
Şekil

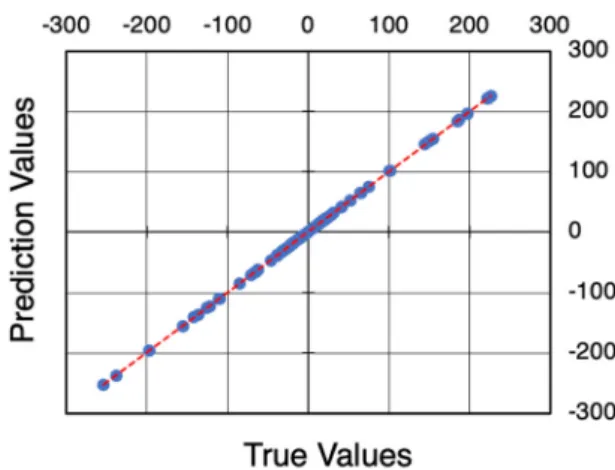
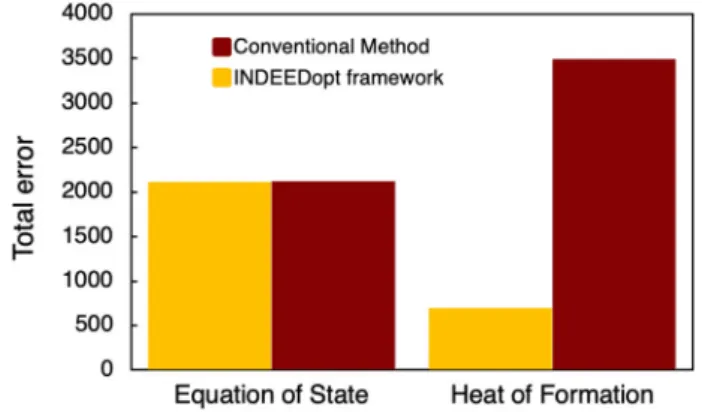

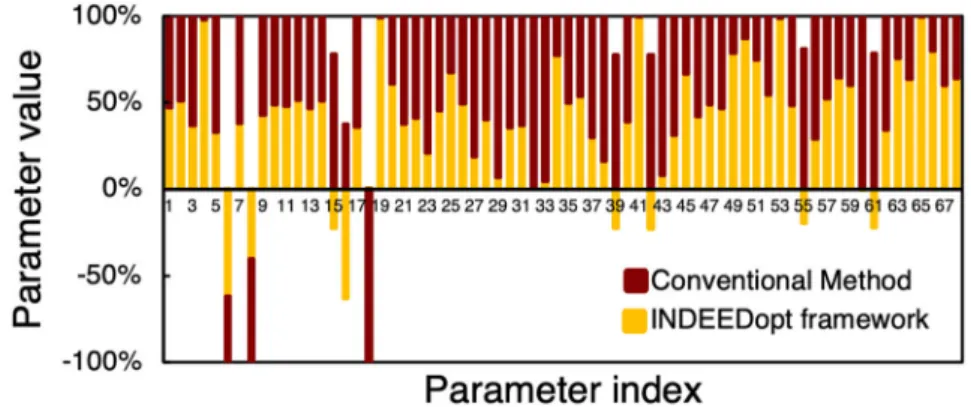
Benzer Belgeler
Bunun birlikte genelde gelişmekte olan ülkelerin gelişmiş toplumlara ekonomik olarak daha ucuza sunduğu turizm, 1950’lerden bu yana küresel bir boyuta ulaşmış
11- Lokal ve sistemik tedavilere cevapların izlenmesi EULAR çalışma grubu tarafından yapılan bir çalış- mada; USG’nin el OA’sında osteofi tleri ve eklem
Nurbanu, tarihe ilişkin çok sayıda inceleme ve araştırması olan Teoman Ergül'ün ilk romanı olarak yayımlandı.. Roman, çok güzel bir cariye olan Nurbanu'nun,
By changing the four diffusion parameters (Ti thickness, waveguide strip width, diffusion temperature, diffusion duration), it is possible to get high polarization
Increasing the pearlite content in the matrix causes to increase the fatigue strength as well as the yield, ultimate tensile strengths and hardness values of ductile iron.. The
Türkçe Dersi Öğretim Programı, bu unsurlara ve millî eğitim sistemindeki değişme- lere (zorunlu eğitim, temel eğitim, ilkokul- ortaokul düzenlemeleri vs.) bağlı
Bizans’ın ve İstanbul’un en önemli limanlarından biri olan Theodosius Limanı’nda elde edilen bu buluntuların ait oldukları dönemin tespiti çözümlenmesi
Transformation matrix for space frames is written according to direction cosines of x, y and z axes of the members in local coordinate system with respect to its global
