In vitro correction of hbb v6g mutation in lymphocyte cells of patients with sickle cell anemia using genome-editing crispr / cas9 technique
Tam metin
Şekil

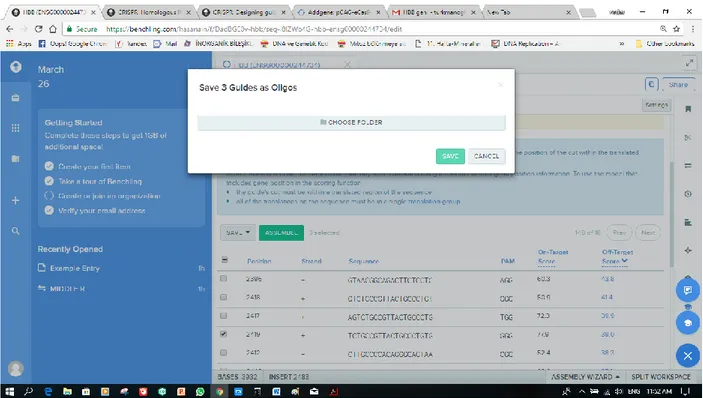
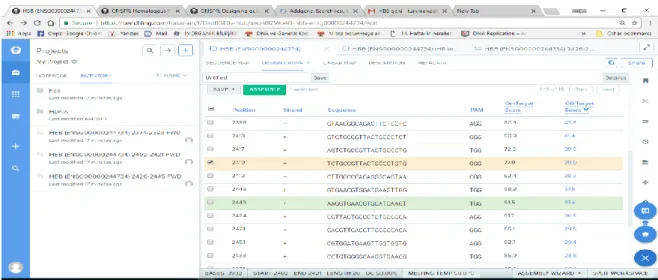
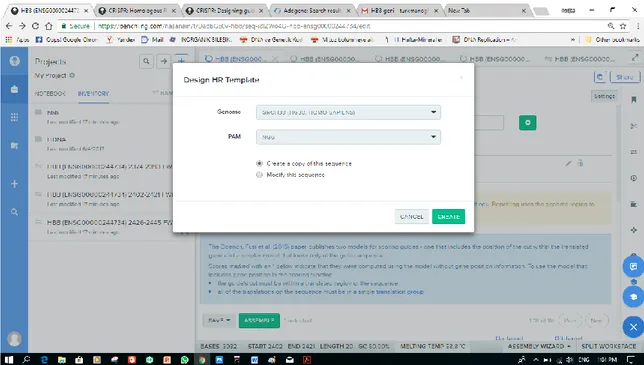
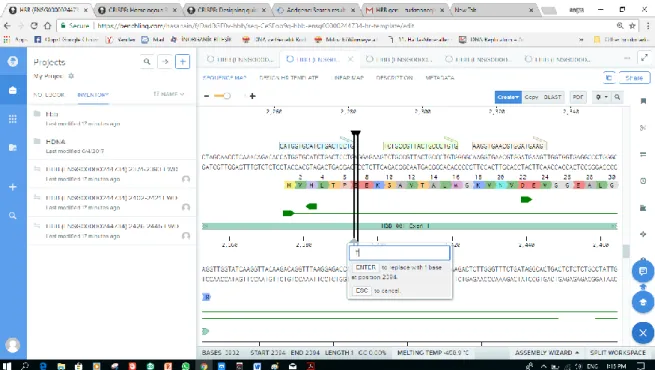
Benzer Belgeler
Bu bulguya göre yaratıcı drama yöntemiyle verilen çevre eğitimi etkinliklerinin uygulandığı deney grubunun çevre eğitimine yönelik farkındalıkları ile çevre eğitimi
ödemeliyim?... 5) Bir oyuncak araba ve bir kalemlik aldım. Ne kadar para üstü almalıyım?... 3) Bir kalemlik, bir oyuncak araba ve bir de silgi aldım. Kasaya 200TL verdim. Kaç TL
The purpose of our study was to investigate the effects of the competitive flow, by measuring both the volume and velocity in the jugular vein bypass grafts, placed in the
The plague of bogus news makes absence of trust in reporting as well as choppiness in political world. Counterfeit news impacts individuals' choices with respect
In our study, we aimed to determine the anemia prevalence and the causes that affect anemia in patients with DM with normal renal function.. Materials and Methods:
Bu bilgiler ışığında Jinek ve ark., CRISPR Tip II sisteminde, hedef DNA’nın özgül ola- rak tanınması için; RNase III’ün gerekli olmadığını, Cas9, tracrRNA
In another study Taşyenen (26) compared the prevalence of anemia in children of primary school educated mother and higher educated mother in Manisa city and
Evaluation of the apoptosis- and necrosis-inducing potentials of CoFe 2 O 4 -NPs as assayed by Annexin V-FTIC apoptosis detection assay with propidium iodide, Results are
