i
T.C.
EGE ÜNĠVERSĠTESĠ TIP FAKÜLTESĠ
ÇOCUK SAĞLIĞI VE HASTALIKLARI ANABĠLĠM DALI PROF. DR. SAVAġ KANSOY
KOLESTEROL ESTER DEPO HASTALIĞININ
OLASI ÖN GÖSTERGELERĠNĠN
ARAġTIRILMASI VE LĠZOZOMAL ASĠT
LĠPAZIN REFERANS ARALIĞININ
BELĠRLENMESĠ
UZMANLIK TEZĠ
DR. BEYHAN ÖZKAYA
TEZ DANIġMANI
DOÇ. DR. SEMA KALKAN UÇAR
ii ÖNSÖZ
Çocuk Sağlığı ve Hastalıkları uzmanlık eğitimim süresince bilgi ve deneyimleri ile eğitimime katkıda bulunan başta Ege Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Başkanı Prof. Dr. Savaş KANSOY olmak üzere, değerli öğretim üyelerine, kıdemlilerime ve uzmanlarıma teşekkür ederim.
Tez çalışmalarım sırasında desteğini ve güvenini benden esirgemeyen; tecrübe, bilgi ve sevgisini benimle paylaşan sevgili hocam, tez danışmanım Sn. Doç.
Dr. Sema KALKAN UÇAR’ a teşekkürü bir borç bilirim.
Tezimin oluşturulması ve hazırlanmasındaki katkı ve yardımları için Sn. Prof.
Dr. Mahmut ÇOKER’e, Sn. Prof.Dr. Eser SÖZMEN’e, Metabolizma BD uzmanları Uzm. Dr. Ebru CANDA’ya , Uzm. Dr. Mehtap KAĞNICI’ya , Uzm.Dr. Melis KÖSE’ye , Uzm. Dr. Havva YAZICI’ya, Uzm. Dr. Esra BEN’e metabolizma
laboratuvar çalışanları Aylin Akın ve Sündüz Ekinci’ye, çalışmaya katılan çocuklarımız ve ailelerine teşekkürlerimi sunarım.
Dört yıl boyunca birlikte çalıştığım, başta Dr. Derya AYDIN, Dr. Müşerref
KASAP ve Dr. Asuman ÖZEN olmak üzere, arkadaşlıklarını ve dostluklarını
paylaştığım çok değerli asistan arkadaşlarıma teşekkür ederim.
Bugünlere gelmemde sonsuz emeği olan, büyük fedakarlıklarla maddi-manevi desteğini esirgemeden beni büyütüp okutan sevgili annem ve babama, her zaman yanımda olan biricik kardeşime ve ablam Aysun YILMAZ’a teşekkür ederim.
İZMİR Dr. Beyhan ÖZKAYA Haziran, 2016
Bu çalışma EÜTF Araştırma Projeleri Alt Komisyonu tarafından desteklenmiştir. Proje no: 2014-TIP-016
iii ÖZET
KOLESTEROL ESTER DEPO HASTALIĞININ OLASI ÖN GÖSTERGELERĠNĠN ARAġTIRILMASI VE LĠZOZOMAL ASĠT
LĠPAZIN REFERANS ARALIĞININ BELĠRLENMESĠ
GiriĢ ve Amaç: Kolesterol ester depo hastalığı nadir görülen, otozomal
resesif kalıtılan lizozomal depo hastalığıdır. Hücre içi triaçilgliserol ve kolesterol esterlerin hidrolizinden sorumlu lizozomal asid lipaz (LAL) enziminin eksikliği veya yetersizliği sonucu oluşur. Sunulan çalışmanın birinci amacı klinik şüphe (hepatik disfonksiyon ve/veya dislipidemi varlığı) olan olgularda LAL eksikliğinin araştırılmasıdır. LAL aktivitesi düşük saptanan olgularda bu düşüklüğün bağlantılı olabileceği biyobelirteçleri belirlemek ve LAL için Türk popülasyonuna ait referans aralığını belirlemek, çalışmamızın ikincil amaçlarıdır. Araştırılan biyobelirteçlerin başında lizozomal hastalıkların tanı ve izleminde kullanılan “kitotriozidaz”, non-alkolik steatohepatitde apoptozisten sorumlu “cytokeratin-18”, ateroskleroz ve non alkolik steatohepatitte değerli bir gösterge olan hsCRP, ve lipid peroksidasyonunda önemli rolleri olan TBARS ve MPO ile ilişkinin araştırılması planlandı.
Gereç ve Yöntem: Çalışmaya 0- 18 yaş aralığında toplam 360 çocuk
alındı. 40‟ı kontrol; 320‟si hepatomegali(+/-), transaminaz yüksekliği(+/-) ve/veya hiperlipidemi saptanan non-obez tarama olguları idi. Bu hastalardan LAL aktivitesi <0.6 nmol/ml/saat Grup 1‟i oluşturdu; LAL aktivitesi ≥0.6 nmol/ml/saat Grup 2‟i meydana getirdi (Şekil 1). Olguların yaş, cinsiyet, ailede hiperlipidemi öyküsü gibi demografik verileri ve fizik muayene bulguları kaydedildi. Hasta dosyalarından lipid profili, hemogram, biyokimya, koagulasyon parametreleri, abdomen USG ve viral seroloji (HbsAg, Anti Hbs, Anti HCV) sonuçları kaydedildi. LAL aktivitesi çalışılması için kapiller kuru kan örneği; LAL aktivitesi düşük saptanan olgularda kitotriosidaz, hsCRP, cytokeratin-18, MPO ve TBARS çalışılması için eş zamanlı 2 cc düz tüpe kan ayrıldı. Referans enzim olarak sfingomyelinaz çalışıldı. LAL enzim düzeyi enzim inhibisyon (Lalistat 2) yöntemi ile çalışıldı.
iv
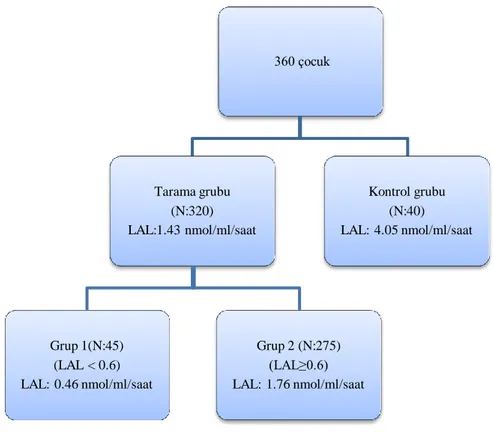
ġekil 1: Çalışmaya alınan kontrol grubun ve tarama hastaların dağılımı ve ortanca LAL düzeyleri
Bulgular: Çalışmaya katılan tüm olguların yaş ortancası 5 ( 0-18) yıl
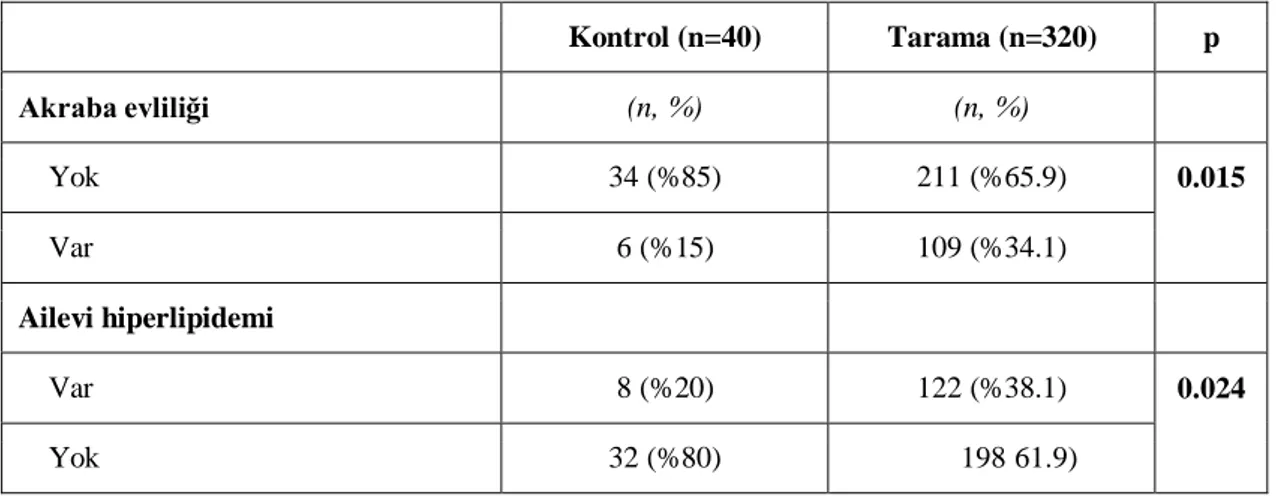
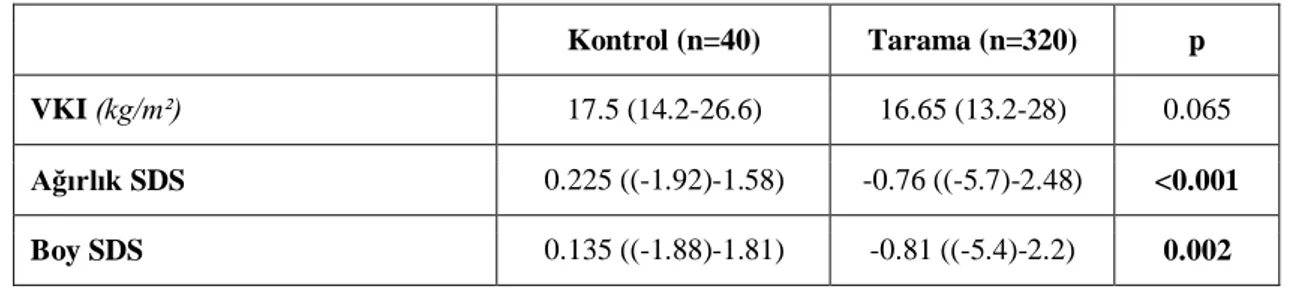
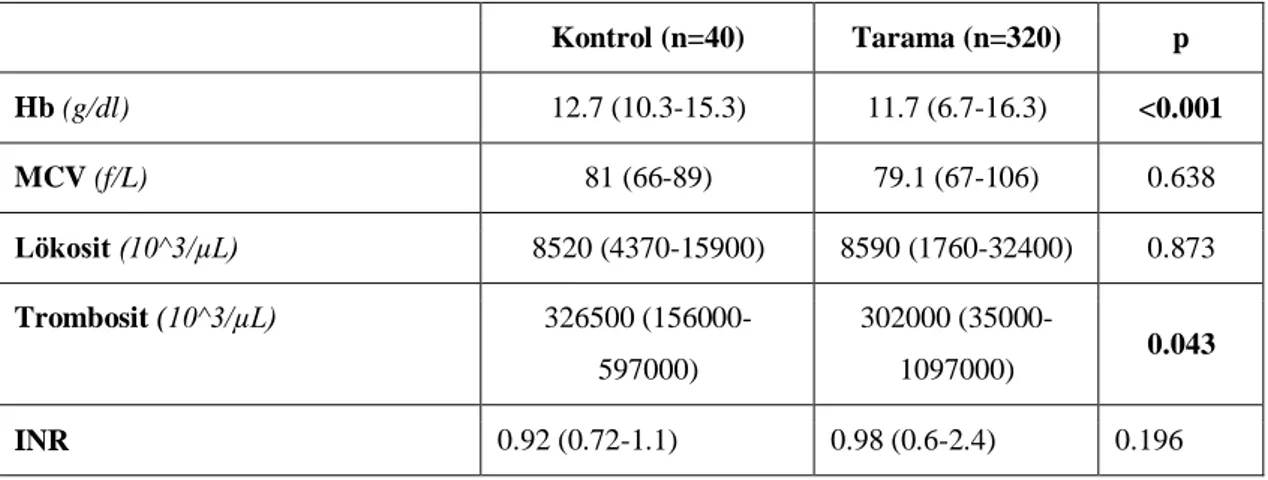
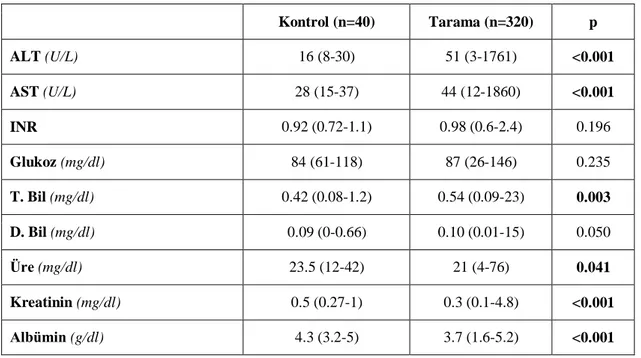
saptandı. Bu olguların 154 (%42)‟ü erkek, 206 (%58)‟sı kız idi. Tarama grubunda akraba evliliği ve ailesel hiperlipidemi anlamlı yüksek saptandı (p<0.05). Boy- ve ağırlık- SDS ortancaları tarama grubunda anlamlı düşük bulundu (p<0.05). Tarama grubunda LAL düzeyi kontrol grubu ile karşılaştırıldığında anlamlı düşük bulundu (p<0.001), fakat LAL eksikliği (LAL<0.03 nmol/ml/saat) olan olgu saptanmadı. Kontrol ve tarama grupları karşılaştırıldığında hemoglobin ve trombosit parametreleri tarama grubunda anlamlı düşük saptandı (p<0.05). Beklendiği gibi tarama grubunda AST, ALT, total bilirubin düzeyi anlamlı yüksek bulundu (p<0.05). Grup 1 ile kontrol grubu karşılaştırıldığında Grup 1‟de ailevi hiperlipidemi öyküsü, ALT, AST ve total bilirubin anlamlı yüksek; Hb, lökosit ve trombosit sayıları anlamlı düşük bulundu (p<0.05). Ayrıca, Grup 1‟de LDL-kolesterol ve trigliserid anlamlı yüksek, HDL-kolesterol anlamlı düşük saptandı (p<0.05). Kontrol ve Grup 1 TBARS düzeyleri (sırası ile: 11.25 (3.75-49.38),14.38 (5-37.5) mmol/ml) anlamlı olarak farklı bulundu (p=0.035). Diğer parametrelerle ilişki saptanmadı. Ayrıca, hepatomegalisi olanlarda hastalarda ortanca LAL aktiviteleri anlamlı olarak düşük
360 çocuk Kontrol grubu (N:40) LAL: 4.05 nmol/ml/saat Tarama grubu (N:320) LAL:1.43 nmol/ml/saat Grup 2 (N:275) (LAL≥0.6) LAL: 1.76 nmol/ml/saat Grup 1(N:45) (LAL < 0.6) LAL: 0.46 nmol/ml/saat
v saptandı (p=0.013). Tüm çalışma grubu değerlendirildiğinde: LAL düzeyi ile LDL, trigliserid ve ALT arasında negatif korelasyon saptandı. Ayrıca ALT ve trigliserid düzeylerinin sırası ile 1.8 ve 1.4 kat artışları LAL aktivitesinde anlamlı bir düşüşe neden oldukları görüldü. Kadın ve erkek için aynı olmak üzere referans aralık 0.26-6.2 nmol/ml/saat olarak belirlendi.
Sonuçlar:
KEDH tanısı alan olgu tanımlanmadı.
LAL düşüklüğünün ALT, LDL-kolesterol ve trigliserid ile negatif korelasyon gösterdiği saptandı.
LAL düşüklüğü TBARS ile ilişkili bulundu.
Eve gidecek mesaj:
Pediatrik yaş grubunda dislipidemi ve/veya transaminaz yüksekliği LAL enzim aktivitesinde düşüklük ile birliktedir.
Anahtar Kelimeler: Lizozomal Asid Lipaz, Kolesterol Ester Depo Hastalığı,
vi SUMMARY
STUDYING THE POTENTIAL PRE-INDICATORS OF
CHOLESTEROL ESTER STORAGE DISEASE AND DETERMINING THE REFERENCE RANGE OF LYSOSOMAL ACID LIPASE
Introduction and Aim: Cholesterol ester storage disease is a rare, lysosomal storage disease with autosomal recessive inheritance. It results from the deficiency or insufficiency of lysosomal acid lipase (LAL) enzyme in charge of the hydrolysis of intracellular triacylglycerol and cholesterol esters. The initial objective of the present study is to search for LAL deficiency in the cases with clinical suspicion (hepatic dysfunction and /or existence of dislipidemia). In the cases determined to have low LAL activity, the secondary objectives of our study are to determine the biomarkers with which this lowness will be associated and to determine the reference range of Turkish population for LAL. At the lead of the searched biomarkers, it was planned to search the relationship with “kitotriozidaz” used in the diagnosis and follow up of lysosomal diseases, with “cytokeratin-18” responsible for apoptosis in non-alcoholic steatohepatitis, with hsCRP which is a valuable marker in atherosclerosis and non-alcoholic steatohepatitis, and with TBARS and MPO which have important roles in lipid peroxidation.
Material-Methods: A total of 360 children were included in the study with the age range of 0-18. 40 of them were included in the control group and other 320 were the non-obese scanning cases diagnosed with hepatomegaly (+/-), transaminase elevation (+/-) and/ or hyperlipidemia. Of these patients, those with LAL activity of <0.6 nmol/ml/hour formed Group 1; those with LAL activity of ≥0.6 nmol/ml/hour formed Group 2. The cases demographic data such as age, gender, family history of hyperlipidemia and results of physical examination were recorded. From the patient files, the results of lipid profile, hemogram, biochemistry, coagulation parameters, abdomen USG and viral serology (HbsAg, Anti Hbs, Anti HCV) were recorded. To study LAL activity, capillary dry blood sample was sorted out; in cases with low LAL activity blood was simultaneously sorted out into 2 cc plain tube to study kitotriosidasis , hsCRP, cytokeratin-18, MPO and TBARS. Sfingomyelinaz was studied as reference enzyme. LAL enzyme level was studied with enzyme inhibition (Lalistat 2 ) method.
vii
Results: Average age of all cases in the study was found to be 5 years (0-18).
154 of the cases (42%) were male and 206 of them (58%) were female. In the scan group, kin marriage and familial hyperlipidemia were found to be significantly high (p<0.05). Height and weight SDS averages were found to be significantly low in the scan group (p<0.05). LAL level in the scan group were found to be significantly low in comparison to control group (p<0.001), yet no case with LAL deficiency (LAL < 0.03 nmol/ml/hour) was found. When the control and scan groups were compared, hemoglobin and thrombocyte parameters were found to be significantly low in scan group (p<0.05). As might be expected, AST, ALT total bilirubin levels were found to be significantly high in scan group (p<0.05). When Group 1 was compared to control group, family history of hyperlipidemia, ALT, AST and total bilirubin were found to be significantly high ; Hemoglobin, leukocyte and platelets numbers were found to be significantly low in Group 1 (p<0.05). Also in Group 1, LDL-cholesterol and trigliserid were found to be significantly high, HDL-cholesterol to be significantly low (p<0.05). TBARS levels of Control group and Group 1were found to be significantly different (11.25 (3.75-49.38),14.38(5-37.5) mmol/ml, respectively) (p=0.035). No relationship was found with other parameters. Besides, in patients with hepatomegali, LAL activities were found to be significantly low (p=0.013). when the whole study group was assessed, a negative correlation was determined between LAL level and LDL, and between trigliserid and ALT. Also, it was seen that 1.8 and 1.4 times increase of ALT and trigliserid levels respectively caused a significant decrease in LAL activity. Reference range was determined to be 0.26-6.2 nmol/ml/hour, as being the same for males and females.
Conclusion:
No patient with KEDH diagnosis was determined.
LAL lowness was found to show negative correlation with ALT, LDL-cholesterol and trigliserid.
LAL lowness was found to be related to TBARS.
The message to home: Dyslipidemia and /or transaminase elevation is together with LAL enzyme activity lowness in pediatric age group.
viii Key words: lysosomal acid lipase, Wolman disease, cholesterol ester storage disease, chitotriosidase, CK-18, hsCRP, MPO, TBARS
ix ĠÇĠNDEKĠLER ÖNSÖZ ... ii ÖZET ... iii SUMMARY ... vi İÇİNDEKİLER ... ix TABLOLAR DİZİNİ ... xi ŞEKİLLER DİZİNİ ... xii KISALTMALAR ... xii 1. GİRİŞ ve AMAÇ ... 1 2. GENEL BİLGİLER ... 3
2.1. Kolesterol Ester Depo Hastalığı/Wolman Hastalığı ... 3
2.1.1. Tanım... 3
2.1.2. Sıklık ... 4
2.1.3. Etyopatogenez ... 4
2.1.4. Klinik ... 7
2.1.5. Laboratuvar ve Radyolojik Bulgular ... 9
2.1.6. Tanı ...10 2.1.7. Genetik ...12 2.1.8. Ayırıcı tanı ...13 2.1.9. Tedavi ...14 2.2. Ön Göstergeler ...17 2.2.1. Kitotriozidaz ...17 2.2.2. Sitokeratinler ve CK-18 ...22
2.2.3. C-Reaktif Protein Ve Yüksek Duyarlıklı CRP ( hsCRP) ...23
2.2.4. Myeloperoksidaz (MPO) ...24
2.2.5.Lipid peroksidasyonu ve TBARS ...25
2.3. Referans Aralığının Belirlenmesi ...26
2.3.1. Normal Değer ...26
2.3.2. Referans Değer...26
2.2.3. Referans Aralığı ...27
2.2.4. Referans toplum seçimi ve değerlendirme ...27
3. MATERYAL VE METOD ...29
x 3.2. Yöntem ...30 3.3 İstatiksel Analiz ...31 4. BULGULAR ...33 5. TARTIŞMA ...55 6. SONUÇ ve ÖNERİLER ...62 KAYNAKÇA ...65
EK 1: OLGU RAPOR FORMU………..75
EK 2: ETİK KURUL ONAY BELGESİ………....76
xi TABLOLAR DĠZĠNĠ
Tablo 1: LAL eksikliğinde gözlenen klinik, serum belirteç ve karaciğer
biyopsi bulgularının özeti………...8
Tablo 2: LAL aktivitesi kime baktık?...30
Tablo 3: Gruplara göre yaş ve cinsiyet dağılımı………...35
Tablo 4: Akraba evliliği ve ailesel hiperlipidemi komplikasyonu öyküsü…..35
Tablo 5: Olgu gruplarının antropometrik ölçümleri………36
Tablo 6: Olgu gruplarının hematolojik parametreleri……….37
Tablo 7: Olgu gruplarının lipid profili……….38
Tablo 8: Olgu gruplarının biyokimyasal belirteçleri………..38
Tablo 9: Tarama grubunda LAL düzeyine göre semptom ve klinik……...…41
Tablo 10: Grup 1 ve Grup 2‟nin lipid profili ve transaminaz değerleri…….42
Tablo 11: Kontrol grubu ile Grup 1 demografik ve antropometrik veriler….44 Tablo 12: Kontrol ve Grup 1 hematolojik parametreleri………45
Tablo 13: Kontrol ve Grup 1 lipid profili……….…...46
Tablo 14: Kontrol ve Grup 1 LAL düzeyi, biyokimyasal parametreleri…….46
Tablo 15: Kontrol ve Grup1'de çalışılan ek biyobelirteçlerin karşılaştırılması……….47
Tablo 16: Farklı gruplar ve yaş aralıkları için LAL düzeyleri……..…48
Tablo 17: LAL düzeyinin cinsiyet değişkenine göre incelenmesi……...…...48
Tablo 18: LAL düzeyi ve kronik yorgunluk arasındaki ilişki…………..…...49
Tablo 19: LAL düzeyi ve büyüme geriliği arasındaki ilişki………..………..49
Tablo 20: LAL düzeyi ile ikter arasındaki ilişki………..49
Tablo 21: LAL düzeyi ve hepatomegali arasındaki ilişki………50
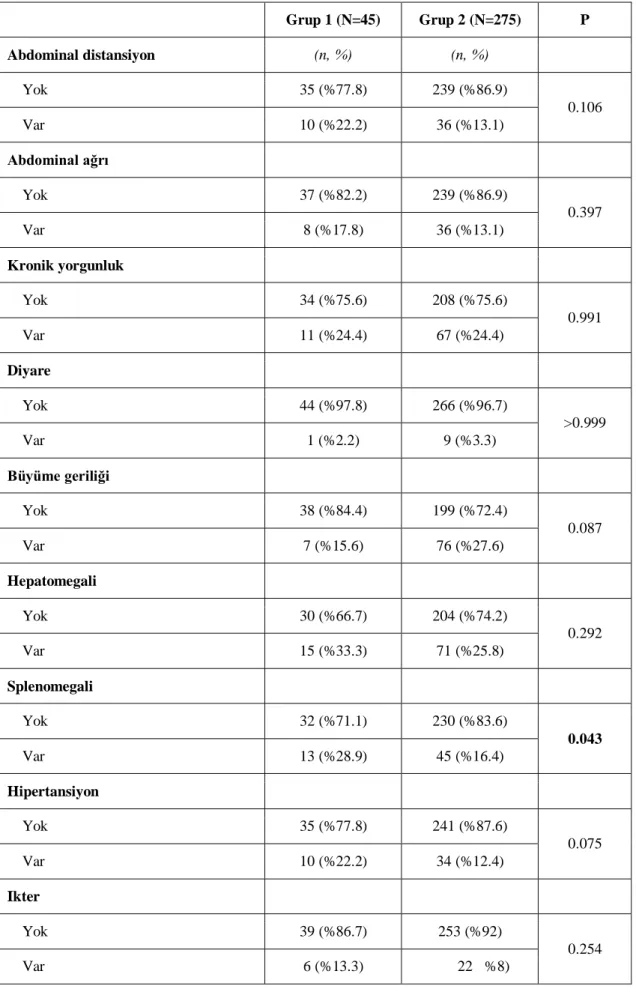
Tablo 22: LAL düzeyi ve splenomegali arasındaki ilişki………50
Tablo 23: Hepatosteatoz USG bulgusu ve değişkenler arasındaki ilişki……51
Tablo 24: LAL ile değişkenler arasındaki korelasyon analizleri……….51
Tablo 25: LAL için “odds‟‟ oranları………53
xii ġEKĠLLER DĠZĠNĠ
Şekil 1: Çalışmaya alınan kontrol grubun ve tarama hastaların dağılımı ve
ortanca LAL düzeyleri ... iv
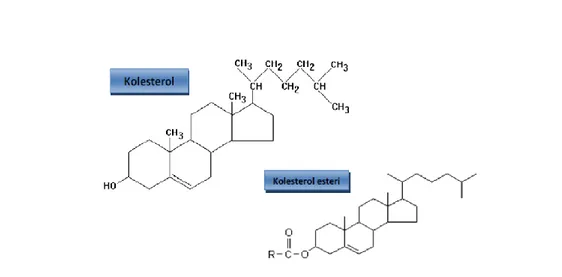
Şekil 2: Kolesterol ve kolesterol esteri ... 5
Şekil 3: Hücresel kolesterol metabolizması ... 6
Şekil 4: LAL eksikliği olan bireyde karaciğer biyopsisi ...10
Şekil 5: Kitotriozidazın yapısı (62) ...19
Şekil 6: Kırılmış CK-18 (71) ...22
Şekil 7: Çalışmaya alınan kontrol ve tarama gruplarının dağılımı ...34
Şekil 8: Kontrol ve tarama grubunun LAL aktivitesi ...37
Şekil 9: Tarama grubunun karın ultrasonografi verileri ...39
Şekil 10: Tarama grubu alt grupları ...40
Şekil 11: Grup 1 ve kontrol grubu dağılımı ...43
Şekil 12: Grup 1 ve kontrol grubunun LAL düzeyleri ...45
Şekil 13: LAL aktivite ve LDL-kolesterol düzeyleri arasındaki ilişki ...52
Şekil 14: LAL aktivite ve Trigliserid düzeyleri arasındaki ilişki ...52
Şekil 15: LAL aktivitesi ve ALT düzeyleri arasındaki ilişki ...52
Şekil 16: LAL referans grubunun histogramı ...54
Şekil 17: Sunulan çalışmamızın Muntoni ve ark. çalışması ile şekilsel olarak karşılaştırılması ...58
xiii KISALTMALAR
LAL : Lizozomal asid lipaz
WH : Wolman hastalığı
KEDH : Kolesterol ester depo hastalığı LĠPA : Lipaz A geni
LDL : Düşük dansiteli lipoprotein HDL : Yüksek dansiteli lipoprotein VLDL : Çok düşük dansiteli lipoprotein HMG-COA: 3-hidroksi-3-metil-glutaril-KoA Apo B : Apolipoprotein B
ABCA1 : ATP-binding cassette transporter 1 NPC1 : Niemann Pick tip C
CK-18 : Cytokeratin 18
hsCRP : Yüksek duyarlıklı CRP PAS : Periyodik asit shift
SREBPs : Sterol düzenleyici eleman bağlayıcı proteinler MPO : Myeloperoksidaz
TBARS : Thiobarbituric Acid Reactive Substance HeFH : Heterozigot familyal hiperkolesterolemi FCH : Ailesel kombine hiperlipidemi
1
1. GĠRĠġ ve AMAÇ
Kolesterol ester depo hastalığı (KEDH) nadir görülen, otozomal resesif kalıtılan lizozomal depo hastalığıdır. Hücre içi triaçilgliserol ve kolesterol esterlerin hidrolizinden sorumlu lizozomal asid lipaz (LAL) enziminin eksikliği sonucu oluşur.
Kolesterol esteri ve trigliserid ağırlıklı olarak karaciğer, dalak ve makrofajlarda birikir. Klinik olarak hepatosplenomegali, hepatosteatoz, transaminazlarda yükselme-hepatik yetmezlik, serum total kolesterol, düşük dansiteli lipoprotein (LDL) kolesterol ve trigliserid seviyelerinde yükselme, yüksek dansiteli lipoprotein (HDL) kolesterolde düşüş, erken ateroskleroz ve mortalite ile kendini gösterir. KEDH tanısı Lipaz A (LIPA) gen mutasyonu, LAL eksikliğinin periferal lökosit ya da fibroblast doku kültüründe gösterilmesi ile konur. Karaciğer biyopsisinde progresif mikroveziküler steatoz, fibrozis, mikronodüler siroz ve hepatosit ve Kupffer hücrelerinde kolesterol ester birikiminin saptanması tanıda yardımcıdır.
LAL eksikliği ile ilişkili iki fenotip tanımlanmıştır. Wolman hastalığı (WH) ağır gidişli, infantil form olup; steatore, şiddetli kusma, büyüme geriliği, hiperlipidemi ve hepatosplenomegali ile tanınır. Yaşamın ilk yılında hepatik-adrenal yetmezliğe bağlı mortalitesi yüksektir. LAL aktivitesinin normalin %1‟den azdır. KEDH daha iyi prognozlu formu olup; rezidüel LAL aktivitesi mevcuttur, yaşamın ilk ya da ikinci dekadında bulgular ortaya çıkabilir. Hepatomegali tek farkedilen klinik bulgusu olabilir, etkilenen bireylerde hiperlipidemiye bağlı olarak erken ateroskleroz riski artmıştır. KEDH olgularında LAL aktivitesi normalin %1-12‟si kadardır.
KEDH olgularında diyet, kolesterol ve ApoB sentezini azaltan kolestramin ve statin tedavileri denenmiş; karaciğer yetmezliğine ilerleyen olguların bazılarına karaciğer nakli yapılmıştır. İnsan rekombinant lizozomal asid lipaz alfa (sebelipase alfa) 2015‟ten itibaren bu hastalık için FDA onaylı enzim replasman tedavisidir.
2 KEDH nadir görülen ve şüphelenilmezse kolay atlanılabilen bir hastalıktır. Tedavi edilemediği takdirde kronik karaciğer hastalığı - karaciğer yetmezliği ve hiperlipidemiye bağlı erken ateroskleroza yol açabilmektedir.
Sunulan çalışmada üç temel amaç ve iki ikincil amaç hedeflendi: Bu çalışmanın birinci temel amacı klinik şüphe (hepatik disfonksiyon ve/veya dislipidemi varlığı) olan olgularda LAL eksikliğinin “selektif tarama” adı verilen yöntem ile araştırılmasıdır. LAL eksikliği açısından incelenen hastalarda olası KEDH tanının konulması durumunda erken tedavi olanağının hastaya sunulması ikincil bir amaçtır.
Çalışmanın ikinci temel amacı hastalık yapıcı olmayan fakat düşük olan LAL aktivitesi grubunda “enzimatik düşüklüğünün” bağlantılı olabileceği olası nedenleri araştırmak idi. Bu kapsamda çalşmaya alınan hastaların klinik ve laboratuvar özellikleri ile LAL düzeyi arasında bağlantılar incelendi. Ayrıca
ikincil olarak llizozomal hastalıkların tanı ve izleminde kullanılan “kitotriozidaz”
düzeyi, non alkolik steatohepatitde apoptoziste rol alan “cytokeratin-18”, ateroskleroz ve non alkolik steatohepatitte değerli bir gösterge olan “hsCRP”, ve lipid peroksidasyonunda önemli rolleri olan “TBARS” ve “MPO” ile ilişkinin araştırılması hedeflendi.
Üçüncü temel amaç olarak LAL aktivitesi için Türk populasyonuna ait
3 2. GENEL BĠLGĠLER
2.1. Kolesterol Ester Depo Hastalığı/Wolman Hastalığı
2.1.1. Tanım
KEDH ve Wolman hastalığı LAL eksikliğine bağlı karaciğer, dalak ve diğer organlarda trigliserid ve kolesterol esterlerinin birikmesine bağlı oluşan, nadir görülen lizozomal depo hastalıklarıdır (1). Otozomal resesif kalıtılan bu hastalıklar LIPA genindeki mutasyonlara bağlı oluşmaktadır. Ateroskleroz, kardiyovaskuler hastalık ve erken mortalite ile ilişkilendirilen dislipidemi en sık gözlenen bulgulardır (2). Mikroveziküler hepatostetoz ve transaminaz yüksekliği ile ortaya çıkan ilerleyici kronik karaciğer hastalığı diğer görülen karakteristik bulgusudur (1).
LAL eksikliği kolayca atlanabilir ya da heterezigot ailesel hiperkolesterolemi, non-alkolik yağlı karaciğer hastalığı, kriptojenik siroz gibi yanlış tanılarla hastalar izlenebilir (3).
LAL eksikliği olan bireylerde değişik bulgular oluşabilir ve progresyon hızı değişkendir (1). İnfantlarda hızlı ve progresif başlangıç gösteren WH ilk kez 1956 yılında tanımlanmıştır (4). Birkaç yıl sonra Fredrickson tarafından 12 yaşında bir erkek olguda hiperkolesterolemi, hepatomegali ve karaciğer biyopsisinde kolesterol ester birikimi gösterilmiştir (5). Geç başlangıçlı bu durum kolesterol ester depo hastalığı olarak isimlendirilir. WH ve KEDH 'in başlangıç zamanları farklı olduğu ancak LIPA geninde mutasyon sonucu aynı moleküler patolojiden kaynaklandığı gösterilmiştir. Bu gen lizozomlarda LDL kolesterol içerisinde taşınan kolesterol ester ve trigliseridi hidrolize edip; serbest kolesterol ve serbest yağ asidi oluşumda görev alan lizozomal asid lipazı kodlamaktadır (6). LAL eksikliğinde değişken progresyon hızlarının gözlenmesi hastalığa sebep olan mutasyonların doğası, rezidüel enzim aktivitesinin derecesine bağlıdır (7).
WH olan bebekler tipik olarak yaşamın ilk haftasında bulgu verir; 6-12 ayda multiorgan yetmezliğine bağlı olarak kaybedilir. Klinik belirtiler intrauterin dönemde başlayabilir; prenatal ultrasonografide polihidroamnios, fetal asid saptanabilir (8). Bebeklerde hastalığın bulguları; hepatosplenomegali, abdominal distansiyon, ishal, kusma, malabsorbsiyon sonucu büyüme geriliği ve karaciğer yetmezliğidir. Bu hastalar da karaciğerde kolesterol ester ve trigliserid masif birikimi sonucu fibrozis
4 ve siroz gelişir. Anormal lipid birikimi dalak, adrenal bez, lenf nodları, intestinal mukoza, vaskuler endotel ve iskelet kasında gözlenebilir (9). WH olan olguların %50'sinde direkt karın grafilerinde adrenal kalsifikasyon gözlenmektedir(10).
Çocuk ve erişkinlerde gözlenen LAL eksikliği (KEDH) değişken klinik bulgular gösterir. Kız ve erkeklerde ortalama semptomların başlangıç yaşı 5 yaş olarak belirtilse de; literatürde tanı yaşı 44 yaş erkek ve 68 yaş kadın hastalar gösterilmiştir (1). Lipid profil anormallikleri tüm yaşlarda gözlenebilir. LDL kolesterol plazma seviyerinde yükselme, HDL kolesterol seviyelerinde azalma ateroskleroz ve kardiyovasküler hastalık için risk faktörüdür. Karaciğer disfonksiyonu ve hepatomegali en yaygın bulgusudur (11). Bu fenotipik bulgular LAL eksikliği için non-spesik olup; etyolojisi aydınlatılamamış karaciğer hastalığı durumlarında bu hastalık düşünülmelidir.
2.1.2. Sıklık
KEDH ve WH sıklığını tam olarak bildirmek zor olsada; erken başlangıçlı form olan WH için tahmini prevelans 1/350000 canlı doğumdur. Diyare, masif hepatosplenomegali, malabsorbsiyon, kaşeksi ve adrenal kalsifikasyonla semptom veren bu formda olgular ilk yıl içerisinde kaybedilmektedir. Çocuk ve erişkinde gözlenen KEDH için tahmini prevelansın Kafkas ırkında 1/150000-300000 canlı doğum (9); Alman ırkında 1/40000 canlı doğum olduğu belirtilmektedir (12). Bu grupta klinik bulgu ve semptomlar değişkenlik göstermekle birlikte ateroskleroz ve karaciğer yetmezliği önemli morbidite ve mortalite nedenidir.
2.1.3. Etyopatogenez
Lizozomal asid lipaz lipid metabolizmasında anahtar role sahiptir; lizozomlarda trigliserid ve kolesterol esterlerinin hidrolizinde rol oynar (Şekil 2) LDL-türevi nötral lipidler (kolesterol ester ve trigliserid) LAL tarafından hidrolize edilir; hücresel kolesterol metabolizması için önemli mediyatörler olan serbest kolesterol ve yağ asitleri oluşur (13). Bu lipidler ve okside ürünleri lipogenezis ve kolesterol sentezinde görevli; gen ekspresyonu ile düzenlenen transkripsiyon
5 faktörleri ile etkileşime girer (sterol regulatory element-binding proteins/SREBPs) (14). Normalde intrasellüler serbest kolesterol artışı; LDL reseptörlerinin SREBP-2-aracılı “down regülasyon” una yol açar (hücre içine kolesterol girişinin azalışı ile sonuçlanır), hidroksimetilglutaril-coenzim A (HMG-CoA) redüktazı “feedback” inhibe eder (kolesterol sentezi azalışı ile sonuçlanır), açil-kolesterol açiltransferazı stimüle eder (kolesterol esterifikasyonu artışı ile sonuçlanır). Aynı zamanda intrasellüler yağ asidi artışı; yağ asid sentezinin SREBP-1c-aracılı “down regülasyonuna” bağlı trigliserid ve fosfolipidin üretimine yol açar (15) (Şekil 3).
ġekil 2: Kolesterol ve kolesterol esteri
Lizozomal asid lipaz aktivitesi yokluğu ya da azlığında kolesterol ester ve trigliserid hidrolize edilemez ve lizozomlarda birikmeye başlar. İntrasellüler serbest kolesterolün azalması HMG-CoA ile endojenöz kolesterol üretiminin ve LDL reseptörleri ile endositozun SREBP aracılı “up regülasyonuna” yol açar. Aynı zamanda apolipoprotein B (Apo B) sentezinde artış ve çok düşük dansiteli lipoproteinin (VLDL) üretimine sebep olur (16). HMG-CoA redüktazın artan ekspresyonu, serbest kolesterol düzeylerinde bir artışa sebep olan SREBP-2 duyarlı hücre içi kolesterol azalmasının öncelikli sonucudur.
6 ġekil 3: Hücresel kolesterol metabolizması (A) sağlıklı ve (B) LAL eksikliği olan hasta birey
(Z.Reiner et all./Atherosclerosis 235(2014) 21-30)
ACAT, açil kolesterol açil transferaz; CE, kolesterol esterleri; FA, yağ asidi; FC, serbest kolesterol; FFA, serbest yağ asidi; HMG-CoA r, hidroksimetilglutaril-koenzim A redüktaz; LAL, lizozomal asid lipaz; LAL-D, LAL eksikliği; LDL-C, düşük dansiteli lipoprotein kolesterol; LDLR, LDL reseptörü; SREBPs, sterol düzenleyici element bağlayıcı proteinler; TG, trigliserid; VLDL-C, çok düşük dansiteli lipoprotein kolesterol
LAL eksikliğinde görülen HMG-CoA redüktaz “up” regülasyonundan kaynaklanan serbest kolesterol düzeylerindeki artışın etkisi tam olarak anlaşılmamıştır; fakat LDL reseptör aktivitesinin “feedback” inhibisyonuna ve LDL kolesterolün dolaşımdan temizlenmesinde azalışa sebep olmaktadır. LAL‟den yoksun hepotositlerde kolesterol sentezindeki artışlar, kolesterolü karaciğerden transportunun doğal yolu olan VLDL kolesterol üretiminde artışlara yol açar; bu durum artan LDL kolesterol üretimiyle sonuçlanır (17).
LAL eksikliği olan hastalarda statinlerin kullanımı bazı endişeleri ortaya çıkarmaktadır. İlk olarak, statinler karaciğerde kolesterol esterlerinin birikmesine yol açar; karaciğer hasarını düşürmesi olası değildir. Serum transaminazlarında artışa ve karaciğer fibrozunun statin kullanan hastalarda siroza ilerlemesine yönelik kanıtlar vardır (1,18). İkinci olarak, statinlerin kolesterol sentezini azaltması beklenmektedir, böylelikle lipoproteinleri içeren Apo B‟nin sekresyonunu azaltıp; LDL reseptörlerinin ekspresyonunu artırır. Bu da plazma LDL kolesterolünü azaltır, fakat plazma LDL kolesterolünün yüksek reseptör aracılı alımı karşısında, karaciğerde kolesterol esterlerinin lizozomal birikimini hızlandırması beklenmektedir.
LAL eksikliği ile zıt olarak, bir diğer lizozomal lipid depo hastalığı olan Niemann-Pick tip C (NPC)‟de, dolaşımdaki total kolesterol ve LDL kolesterol azalır. NPC‟i olan hastalarda, serbest kolesterol; sterolü sitozolik kompartmana taşımada rol alan NPC proteinlerindeki kusur nedeniyle lizozomlarda birikir (19).
7 LAL eksikliği olan hastalarda tipik olarak, yüksek serum total kolesterol, yüksek LDL kolesterol, düşük HDL kolesterol gözlenir ve ayrıca yüksek trigliserid düzeyleride saptanabilir (11). Total kolesterol ve trigliserid artışı, VLDL kolesterol ve LDL kolesterol gibi ApoB içeren lipoproteinlerin plazmada birikmesinden kaynaklanır. HDL kolesterol seviyesindeki düşüş, en azından in vitro olarak, azalmış matur HDL formasyonuna (α – HDL partikülleri) ve adenosin trifosfat-bağlayan kaset transporter A1(ABCA1)‟in azalmış ekspresyonuna bağlıdır. ABCA1 geninin ekspresyonu, ABCA1 geninin promotörünü etkileyen nükleer karaciğer X-reseptörünün (LXR) oksisterol –bağımlı aktivasyonu ve artmış hücre kolesterol ile indüklenir (20).
2.1.4. Klinik
LAL eksikliği olan hastalarda; başlangıç semptom ve bulguları değişkenlik göstermektedir. Karaciğer, kardiyovasküler ve metabolik hastalıklarda saptanan dislipidemi, hepatomegali, karaciğer hücre hasarlanması sıklıkla LAL eksikliğinde de gözlenir. Serum transaminaz yüksekliği, fibrozis ve siroza gidiş gözlenebilir (Tablo 1).
LAL eksikliği olan infantlarda klinik; çocuk ve erişkin dönemle kıyaslandığında daha gürültülüdür. Kusma, diyare, steatore ve abdominal distansiyon gibi gastrointestinal semptomlarla birlikte büyüme geriliği ilk başvuru bulgularıdır (9). Masif hepatosplenomegaliye bağlı abdominal distansiyon oluşabilir. Çocuk ve infant olguların %50'sinde radyolojik görüntülemelerde karakteristik bulgu olan noktasal kalsifikasyonlar gözlenir (10). Anemi ve geç klinik bulgular olarak multiorgan yetmezliği, sarılık, karaciğer sirozu ve kaşeksi oluşabilmektedir. Santral sinir sistemi etkilenimi yaygın olmamakla birlikte; LAL eksikliğinden ziyade malnutrisyona ve spesifik nutrisyonel eksikliklere bağlı olabileceği düşünülmüş ya da kemik iliği nakli komplikasyonu olarak gözlenmiştir (9).
LAL eksikliği olan çocuk ve erişkinler; infantlardaki gibi gürültülü bir tablo ile gelebileceği gibi insidental olarak da tanınabilir. Örneğin litaratürde anemi ve hepotosplenomegali ile başvuran 36 yaşında bir kadın hasta araştırıldığında, KEDH tanısı almıştır. Tıbbi geçmişi sorgulandığında 2 yaşında sarılık, hepatomegali ve
8 kahverengimsi cilt lekeleri ile hospitalize edildiği öğrenilmiştir; bu bulgular hastalığın erken göstergesi sayılabilir (21). KEDH tanılı çocuk olguların yaklaşık üçte biri diyare, kusma, abdominal ağrı, malabsorbsiyon ve yağlı dışkılama gibi ciddi gastrointestinal semptomlarla başvurabildikleri bildirilmiştir (22). Kolestaz, büyüme geriliği, adrenal kalsifikasyon, safra kesesi disfonksiyonu olabileceği belirtilmiştir.
Tablo 1: Lizozomal asid lipaz eksikliğinde gözlenen klinik, serum belirteç ve karaciğer
biyopsi bulgularının özeti
Klinik Bulgu semptomlar Hepatomegali/hepatosplenomegali Diyare
Abdominal ve epigastrik ağrı Kusma Anemi Malabsorbsiyon Kolestaz Steatore Büyüme geriliği
Safra kesesi disfonksiyonu Koroner arter hastalığı Anevrizma
İnme
Adrenal kalsifikasyon Özofagus varisleri
Serum belirteçleri Total kolesterol yüksekliği
LDL kolesterol yüksekliği HDL kolesterol düşüklüğü Serum transaminaz yüksekliği
Karaciğer biyopsi bulguları Sarı-turuncu renk değişimi
Lipid yüklü hepatosit ve kuppfer hücreleri
Mikroveziküler steatozis
(makroveziküler steatozisde eşlik edebilir) Fibrozis
9
2.1.5. Laboratuvar ve Radyolojik Bulgular
Total plazma trigliserid ve kolesterol düzeyleri çoğu kez LAL eksikliği olan bebeklerde normaldir, fakat düşük plazma HDL kolesterol düzeylerine ek olarak yüksek trigliserid ve VLDL kolesterol düzeyleri bildirilmiştir (23).
LAL eksikliği olan çocuk ve yetişkinlerde çoğunlukla yüksek total kolesterol, yüksek LDL kolesterol, yüksek ApoB ve düşük HDL kolesterol düzeyleri mevcuttur; bu tablo tip IIa veya tip IIb hiperlipidemiye benzer (24). 9 yaşında ölen LAL eksikliği olan bir çocukta aortada aterosklerotik plak saptanmıştır. Dislipidemi, ateroskleroz ve prematüre kardiyovasküler hastalık ile bağlantılıdır (25).
LAL eksikliği olan bebekler; büyüme geriliği, hepatomegali, karaciğer fibrozu ve sirozu ile birlikte hepatosellüler yetmezliğe hızlı ilerler. Dalak da büyüyebilir ve 2-3 aylıkken normal boyutunun 20 katına ulaşabilir (26). Hepatomegali ve splenomegali LAL eksikliği olan çocuklar ve yetişkinlerde sırasıyla yaklaşık %99 ve %74 oranında görülür (27). Serum alanin aminotransferaz (ALT) ve aspartat aminotransferaz (AST)‟ın yüksek değerleri karaciğer hastalığının erken belirteçleridir, değerler çok az yükselebilir ve hastalar arasında değişim gösterebilir. Klinik görünümler, erken çocukluk döneminde bulunabilen ciddi karaciğer hastalığından (örn: özofageal varisler, karaciğer yetmezliği); ileri erişkinlikte sessizce siroza dönüşen asemptomatik kronik karaciğer hastalığına (örn: yüksek transaminaz) kadar değişim gösterebilir.
Biyopside, karaciğer parlak sarı-turuncu renkte görülür; histolojik analizde hepatosit lizozomlarında kolesterol esterleri ve trigliseridlerin birikmesi sebebiyle portal ve perilobular fibroz ve mikrovezikular steatozun değişen dereceleri gözlenir (3). Karakteristik bir özelliği, periyodik asit-Schiff (PAS) ile güçlü bir şekilde boyanan köpüklü bronz-renkli sitoplazmasıyla birlikte belirgin olarak hipertrofik Kupffer hücreleri ve portal makrofajların varlığıdır. Bu gibi vakuollerın membranları PAS diastazı ile iyi boyanır (şekil 4). Alınan biyopsi örneklerinde, kolesterol esterlerinin çift kırılmalı depolanan likid kristallerinin varlığı LAL eksikliğinde ek bir tanısal ipucudur. Lipid damlacıklarının etrafındaki luminal katepsin D ve lizozomal işaretlerinin (lizozom-ilişkili membran proteini [LAMP1, LAMP2] ve lizozom integral membran proteini 2) immünohistokimyasal belirlenmesi de LAL eksikliğinde faydalı göstergelerdir (3). Çeşitli boyutlardaki kolesterol esterleri ve
10 trigliserid damlacıkları çoğu tek bir membran ile çevrelenen parenkimal ve Kupffer hücrelerinde görülebilir. Lipid vakuollerinin bazıları, depolanan materyale güve-yeniği görüntüsü veren, örnek hazırlama sırasında çözünen kolesterol esterlerinin yıldız-benzeri kristalin izlerini içerebilir (9).
İlerleyen lipid birikimi fibroza (çocuklukta veya yetişkinlikte tanı alan bireylerin üçte ikisinde bulunur) ve sonunda mikronodüler siroza sebep olur (3). Siroz ile bağlantılı komplikasyonlar, portal hipertansiyon, asit, özofagus varisi, gastrointestinal kanama, kaşeksi ve komayı içerir ve bunlar çoğu kez karaciğer yetmezliği ve ölümle sonuçlanır (1).
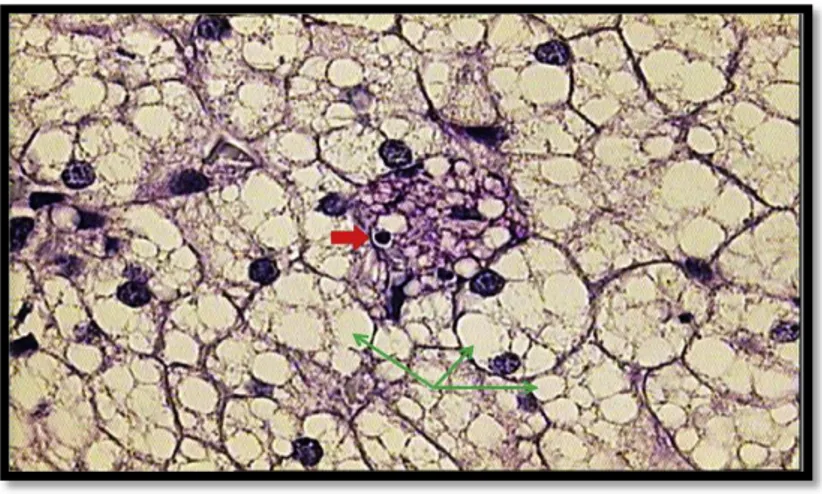
ġekil 4: LAL eksikliği olan bireyde karaciğer biyopsisi (Z.Reiner et all./Atherosclerosis 235(2014)
21-30) Yeşil ok: Hepatositlerde mikroveziküler steatozis, Kırmızı ok: PAS pozitif perivenüler zonda bulunan yağ yüklü Kupffer hücresi
2.1.6. Tanı
LAL eksikliğine yönelik teşhis, yetersiz LAL aktivitesinin veya LIPA genindeki mutasyonların gösterilmesi ile konur (1). Biyopsi bulguları ve radyolojik bulgular LAL eksikliğinde şüphe ve tanı konusunda yardımcı olurlar.
LAL aktivitesinin ölçümü
Kültüre fibrolastlar, periferal lökositler veya karaciğer dokusundaki enzim aktivitesini ölçülerek LAL eksikliği tanısı biyokimyasal olarak doğrulanabilir.
Ancak, bu tahlillerdeki substratlar (örn: 4-nitrofenil palmitat) LAL için spesifik olmayabilir, dolayısıyla yanlış bir negatif sonucun oluşturulması teorik olarak mümkündür. Kuru kandaki LAL aktivitesini belirlemek için yeni bir yöntem geliştirilmiştir ve hastaları saptadığı bulunmuştur (28). Florimetrik substrat
4-11 metilumbeliferil palmitat kullanılarak LAL aktivitesi ölçülür. Tüm kandaki diğer lipazlar kuru kan damlalarındaki LAL ölçümüne karışabilir (29), bu nedenle bir LAL inhibitörü kullanılır. Lalistat 2, NPC1 için olası bir törapatik hedef olarak önceden geliştirilmiş olan oldukça spesifik LAL inhibitörüdür (30). Total lipaz aktivitesini; Lalistat 2 varken lipaz aktivitesi ile karşılaştırarak LAL aktivitesi belirlenir; iki sonuç arasındaki fark LAL enzimine atfedilebilir. Bu metot, orta derecede LAL aktivitesi gösteren taşıyıcılarla birlikte sağlıklı ve hasta bireyler arasındaki farklılığı gösterir.
Genetik Test
LIPA‟nın kodlama alanlarının tam sekanslaması LAL eksikliği olduğundan şüphelenilen bireylerinin genetik durumunun karakterizasyonunu sağlar (9). LAL eksikliği çocuklar ve yetişkinlerdeki mutant alellerin %50-70‟inde en yaygın mutasyon E8SJM mevcut olmasına rağmen, bazı popülasyonlarda ki E8SJM mutasyonunun düşük sıklığı (örn: Afrika Amerikalılar ve Asya kökenli olanlar), polimeraz zincir reaksiyonu tahlilleri yoluyla yaygın mutasyon görüntülemenin bu popülasyonlarda yeterli olmayabileceği anlamına gelmektedir (31).
En çok etkilenmiş hastalar LIPA mutasyonları için homozigot ya da bileşik heterozigot olmasına rağmen, bu hastaların rutin genetik analizinde saptanmayan intronik mutasyonları olabilir. Bunun ışığında, LAL eksikliğini teşhis etmek için sekanslamanın kullanılması kolay ulaşılır, kesin ve düşük maliyetli DBS tahlili ile değiştirilebilir.
Karaciğer biyopsisi
Karaciğer biyopsisi genellikle karaciğer anormalliklerini değerlendirmek için en güvenilir yöntem olarak görülür. Bununla beraber, biyopsi ile bağlantılı morbidite ve mortalite riski ve bu işlemle bağlantılı maliyetler; yaygın olarak uygulanmasını engellemektedir. Örneklem hatası bu yöntemle teşhis yapmayı zorlaştırabilir. Mevcut kılavuzlar karaciğer biyopsisinin, ancak, kan testi gibi diğer invazif olmayan yollarla sonuca ulaşılamazsa teşhis elde etmek için kullanılması gerektiğini ileri sürmektedir (32).
Karaciğer biyopsisindeki mikrovezikuler steatozun varlığı LAL eksikliğine özgü bir durum değildir, dolayısıyla bir tanıyı doğrulamak için diğer histolojik işaretler gerekmektedir. Köpüksü, bronz -renkli sitoplazma ile birlikte hipertrofik
12 Kupffer hücreleri ve portal makrofajlar çocuklar ve yetişkinlerdeki LAL eksikliğinin karekteristik özelliğidir. Lipid vakuolları etrafındaki luminal ve membran lizozomal işaretlerin varlığı, alınan örneklerdeki patognomonik kolesterol ester kristalleri oldukları gibi, parafine gömülü materyaldeki LAL eksikliği göstergesidir (3).
Radyolojik teknikler
3T manyetik rezonans görüntüleme kullanarak yapılan hepatik manyetik rezonans spektroskopi, LAL eksikliği ile bağlantılı hepatik lipid birikimini tanımlamak ve miktarını belirlemek için faydalı invazif olmayan bir yöntem olarak son zamanlarda kullanılmaktadır (33). Bu yaklaşım teşhis ve hastalık izlemi için tekrarlayan biyopsi örneklemi ile kıyaslandığında daha elverişli bir alternatiftir.
2.1.7. Genetik
LAL eksikliği 10q23.2 kromozom bölgesinde bulunan LIPA genindeki mutasyonlara bağlı oluşmaktadır. Bu gen yaklaşık 45 kb uzunluğunda olup; 10 ekson içermektedir(9). Bu hastalığa sebep olan 40'tan fazla mutasyon tanımlanmıştır. LAL eksikliği otozomal resesif kalıtılır. Etkilenen bireylerde tipik olarak homozigot, heterozigot ve bileşik heterozigot mutasyonlar bulunabilir. Çoğu ciddi değişiklikler nonsense, çerçeve kayması ya da bitiş kodonu ile sonuçlanan nokta mutasyonlara bağlı oluşmaktadır ve infant dönemde bulgu vermektedir. Ciddi olmayan diğer mutasyonlar çocuk ve erişkinleri etkilemektedir(33).
En yaygın gözlenen mutasyon ekson 8 splice mutasyondur. E8SJM (c.894G>A) çocuk ve erişkinde gözlenen mutasyonların %50'sini oluşturur (19,58). Bu mutasyon, mRNA‟daki ekson 8‟in delesyonu ile sonuçlanan alternatif bir akseptör eklenme alanı ortaya çıkartır. mRNA‟nın az bir miktarı doğru eklenir ve bu da bir miktar arta kalan LAL aktivitesinin ekspresyonu ile sonuçlanabilir. Genel popülasyona yönelik çalışmalar E8SJM allelinin sıklığının Kafkaslarda/beyazlarda 0.0013 (ABD, 0.0017; Almanya, 0.0025; AB, 0.0012), Birleşik Devletler İspanyollarında /hispaniklerinde 0.0017, BD Eskenazi Yahudilerinde 0.0010, Asyalılarda 0.0005, ve Afrika kökenli Amerikalılarda 0.0000 olduğunu göstermiştir (31,34). LAL eksikliği olan çocuklar ve yetişkinlerin %50-70‟inin E8SJM mutasyonu olduğu varsayımına göre, genel hastalık prevelansının etnik ve coğrafi
13 lokasyona bağlı olarak 1/40.000 ile 1/300.000 arasında olabileceği tahmin edilmektedir (31). Bu tahmin literatürde bildirilen az sayıda LAL eksikliği vakasıyla çelişmektedir, bu da hastalığın özellikle Avrupalı olan hastalarda yetersiz teşhis edildiğini göstermektedir (1). Avrupa‟da LAL eksikliğinin insidansına yönelik hiçbir resmi çalışma yapılmamıştır. Avustralya‟da 700.000‟de 1‟den daha az sıklık bildirilmiştir (35). Irak veya İran kökenli Yahudi bebeklerin Los Angeles topluluğunda 1/4200 tahmini insidansı ile en yüksek LAL eksikliği risk grubunda oldukları görülmektedir (36).
2.1.8. Ayırıcı tanı
Diğer kardiyovasküler, karaciğer ve metabolik hastalıklara benzerliği sebebiyle, LAL eksikliğinin tanısını koymak zorlayıcı olabilir. Doğru tanı konmazsa, bu benzerlikler yanlış teşhise ve tedavide gecikmeye neden olabilir (1).
LAL eksikliğindeki serum lipid seviyeleri değişiklik gösterebilir, fakat çoğu çocuk ve yetişkin yüksek total kolesterol ve LDL kolesterol, ve düşük HDL kolesterol değerleri gösterir. Ayırıcı tanı heterozigot familyal hiperkolesterolemi (HeFH) , ailesel ApoB bozukluğu, ailesel kombine hiperlipidemi (FCH) ve poligenik hiperkolesterolemi‟yi kapsamaktadır (37). Birkaç basit prensibe bağlı bu durumlardan LAL eksikliğini ayırmak mümkündür. İlk olarak, detaylı bir soy geçmişi otozomal baskın bozuklukları (örn: HefH, FCH, ve poligenik hiperkolesterolemi) otosomal resesif bozukluklardan (örn: LAL eks.) ayırabilir. İkinci olarak, LAL eksikliğindeki total kolesterol ve LDL kolesterol değerleri; HeFH‟deki kadar yüksek olmayabilir. Ek olarak, HDL kolesterol değerleri LAL eksikliğinde; HeFH‟dekine kıyasla daha düşüktür (38). Benzer lipid profilleri görülebildiğinden; hekimler teşhis olarak HeFH‟yi düşürken LAL eksikliği olasılığını da akla getirmelidirler.
LAL eksikliğinde gözlenen karaciğer anormallikleri iyi karakterize edilmesine rağmen, karaciğer hastalığının ilerleme hızı ve bu nedenle başlangıç semptomları değişkendir. Örneğin, çoğu hastada hepatomegali görülür. Ilımlı yükselmiş veya nadiren normal serum transaminaz değerleri eşlik edebilir. Hepatomegali ve sürekli yüksek serum transaminazları olan hastalarda yanlış tanılar
14 non alkolik yağlı karaciğer hastalığı (NAFLD) , non alkolik steatohepatit NASH) veya kriptojenik karaciğer hastalığını kapsayabilir (1). Viral hepatit veya otoimmün karaciğer hastalığı gibi daha yaygın hastalıkları elemek için viral/immünolojik profil bakılmalıdır (39). Dislipidemi, yağlı karaciğer ve yüksek karaciğer transaminazlarla da ilişkili olan metabolik sendromu olan hastaların tersine, LAL eksikliği olan hastalar obez olmayabilir. Bu nedenle, bu karaciğer görünümleri olan obez olmayan hastalarda LAL eksikliği düşünülmelidir.
2.1.9. Tedavi
LAL eksikliği olan hastalar için hastalığa özgü tedavi son yıllara kadar bulunmamaktaydı. Hastalık komplikasyonlarını hafifletmek için varolan yaklaşımlar destek tedavilerine yönelikti. LAL eksikliği olan hastalardaki temel bozukluğu hedefleyen rekombinant insan lizozomal asit lipaz olan sebelipaz alfanın güvenliğini ve verimliliğini araştırmak için klinik deneyler tamamlanmış olup günümüzde bu hastalık için enzim replasman tedavisi uygulanmaktadır.
Lipid düĢüren terapiler
Statinler (HMG-CoA- redüktaz inhibitörleri), kardiyovasküler hastalık riskini düşürdüğü bilinen, iyi tolere edilen LDL kolesterol düşüren ajanlardır (40). LAL eksikliği olan çocuk ve yetişkinlerde, monoterapi olarak ya da diğer lipid düşüren ilaçlarla beraber statinlerin literatürde bildirilen pek çok vakada LDL kolesterolü düşürdüğü bulunmuştur, fakat bazı hastalarda LDL kolesterol yine de yüksek olduğu raporlanmıştır (41). Yakın zamanda yapılan bir çalışma, LAL eksikliği olan pek çok hastada lipid- düşüren terapilerle yapılan tedavilere rağmen dislipideminin sürdüğüne dikkat çekmiştir (18). LAL eksikliği olan bazı hastalarda statin terapisi ile fibroblastlardaki endojenöz kolesterol sentezinde anlamlı azalma gösterilmiştir (17). Bir başka çalışmada, dolaşımdaki LDL kolesteroldeki düşüşün ApoB içeren lipoproteinlerin azalmış hepatik üretiminden kaynaklandığı sonucuna varılmıştır (42). Statin terapisinin plazma LDL kolesterol değerini düşürebilmesine ve kardiyovasküler riski azaltabilmesine rağmen, LAL eksikliği olan hastalarda hepatik hasarın ilerlediğine dair kanıt vardır (42). Statinlerle tedavi edilen LAL eksikliği olan bazı hastalarda karaciğer boyutunda küçülme olduğu bildirilmiştir; ancak, takip
15 edilen tüm bireylerde karaciğer fibrozu ilerlemeye devam etmiştir (43). Benzer olarak, yapılan bir inceleme statin ile tedavi edilen 12 hastada karaciğer histolojisinin iyileşmediğini açığa çıkarmıştır (1). Tekrarlanan biyopsilerde 12 hastanın tamamında öncekine göre daha ilerlemiş olan progresif karaciğer hastalığı görülmüş; bu da LAL eksikliğindeki karaciğer hastalığının progresif doğasını göstermiştir. Statinle tedavi edilen altı hasta transplantasyona gerek duymuştur veya karaciğer yetmezliğinden ölmüştür (1).
Ezetimibin (kolesterol absorpsiyon inhibitörü ) 6 aylık tedaviden sonra LAL eksikliği olan 15 yaşında bir erkek çocukta karaciğer transaminazlarını normalleştirdiği ve total kolesterol ve LDL kolesterol değerlerini düşürdüğü (sırasıyla %30 ve %25) bildirilmiştir. Bazalde yüksek olan sitokinlerin ve oksidatif stres parametrelerinin serum seviyelerinin de 1 yıl ezetimibe terapisinden sonra normalleştiği bulunmuştur(44).
E vitamini
Bir in-vitro çalışmada, tokoferolün lizozomal ekzositozu uyardığı ve böylece NPC1 ve LAL eksikliği olan bebeklerden alınan fibroblastlarda lipid birikmesini azalttığı bulunmuştur (45). D-tocopherol vitamin E derivesi olup, plazma konsantrasyonu <1mm'dir ve P450 enzim CYP4F2 tarafından hızla oksidasyona uğrar; bu yüzden insan ve hayvan modellerinde yeterli konsantrasyon düzeyine erişilememiş; kullanmı kısıtlanmıştır.
Kök hücre ve karaciğer nakli
Kök hücre nakli LAL eksikliği olan birkaç bebekte gerçekleştirilmiştir (46). Bu yaklaşım hastalığın çok sistemli doğasına yönelmede sınırlı başarıya sahip olmuştur. Takip verileri, karaciğer naklinden 5 yıl sonrasında kadar LAL eksikliği olan bazı hastalarda sağkalım göstermektedir, fakat pek çoğu için uzun vadeli sonuçlar ve diğer eşzamanlı hastalıklar konusunda sınırlı bilgi vardır (47).
Enzim replasman tedavisi
Enzim replasman tedavisi (ERT) diğer lizozomal hastalıklarda başarılı bir şekilde kullanılmaktadır ve LAL eksikliği olan hastalar içinde uygulanmaktadır (76). LAL eksikliği olan hastalar için ERT‟nin hedefi kolesterol esterlerinin ve
16 trigliseridlerin birikmesini engellemek için yakın fizyolojik enzim değerlerini elde etmek ve bunun sonucunda normal organ işlevini sürdürmektir.
Rekombinant insan LAL enzimi olan sebelipaz alfanın Faz III klinik deneyleri sonuçlanmakla beraber; LAL eksikliği olan 9 yetişkinde, faz II çalışması yapılmış olup (LAL-CL01) ve bu hastaların 7‟sini içeren uzun dönem infüzyon (LAL-CL04) verileri diğer bir çalışmada bildirilmiştir (48,49). İlk çalışmada, hastalara 4 hafta süreyle; haftada bir 1 ya da 3 mg/kg sebelipase alfa infüzyonu verilmiş; bunu iyi tolere ettikleri gözlenmiştir. 4 haftanın sonunda karaciğer transaminazlarında hızlı düşüş gözlenirken ve total kolesterol, LDL kolesterol ve trigliseridlerin serum düzeylerinde artış saptanmıştır. Bu durum dokularda birikmiş lipid mobilizasyonunu ortaya koymaktadır (49). LAL-CL01 çalışması tamamlandıktan ve hastalar sebelipase alfayı tükettikten sonra, hem karaciğer enzimleri hem de lipid değerleri taban değerlerine dönmüştür. Kapsamlı çalışmaya tekrar kaydolan hastalara, uzun vadeli iki haftada bir infüzyona (1veya 3 mg/kg) geçmeden önce dört kez haftada bir sebelifaz alfa infüzyonu verilmiş. Yedi hasta sebelifaz alfa ile 78 haftalık tedaviden sonrası değerlendirildiğinde, ALT ve AST taban değere kıyasla düşmeye devam ederek; normal değerlere geldiği gözlenmiştir. Ek olarak, lipid düşürücü ilaç kullanan bu hastalar için sebelipase alfa tedavisi; sırasıyla LDL kolesterol ve trigliserid değerlerinde ortalama %52 ve %40 düşüş ve HDL kolesterolde ortalama %37 artış sağlamıştır (50). Deney sonuna kadar 250‟den fazla infüzyon uygulanmış ve uzun vadeli dozlarda hiçbir güvenlik sorunu ortaya çıkmamıştır. İlaçla ilgili hiçbir ciddi yan etki bildirilmekle birlikte; yan etkilerin çoğu hafif ve enzim ile alakasız; hafif gastrointestinal olaylar (diyare, abdominal kramp) olarak raporlanmıştır (50).
LAL eksikliğinde kullanılan sebelipaz alfanın etkinliği ve güvenilirliği, randomize, çift-kör, plasebo- kontrollü, Faz III çalışması ile değerlendirilmiştir ( ARISE deneyi; ClinicalTrials.gov identifier : NCT01757184). LAL eksikliği olan olgulara 1 mg/kg sebelipase alfa (N=36) ya da plasebo (N=30) 20 hafta boyunca; 2 haftada bir olacak şekilde verilmiş (11 infüzyon). Çalışmaya katılan olguların ALT düzeyleri normalin 1.5 katından daha yüksek, hastaların %58‟sinde LDL>190 mg/dl, karaciğer biyopsisi yapılmış olan olguların tamamında fibrozis ve olguların %31‟inde siroz mevcut olarak belirtilmiş. Çalışmanın sonuçları değerlendirildiğinde ALT değerlerinin sebelipase alfa uygulananların %31‟inde; plasebo uygulananların
17 %7‟sinde normale döndüğü gözlenmiştir (51). Ayrıca sebelipase alfa uygulanan grup, plasebo uygulanan grupla karşılaştırıldığında; AST, LDL kolesterol, trigliserid ve hepatik yağ fraksiyonunda bazal değerlere göre azalma olduğu; HDL kolesterolde bazal değerlere göre artış olduğu gösterilmiştir.
Çok-merkezli Faz II-III (ClinicalTrials.gov identifier: NCT01371825) çalışması dahilinde büyüme geriliği gösteren, LAL eksikliği saptanan (Wolman hastalığı) ve çalışmayı kabul eden 9 bebeğe (ortalama yaş:3 ay, 1.1-5.8 ay aralığında) 0.2-0.35 mg/kg doz başlanıp; 1-3 mg/kg/doz haftalık sebelipase alfa tedavisi uygulanmış. Olguların tümünde büyüme geriliği, hepatosplenomegali, diyare, kusma, abdominal distansiyon, karaciğer yetmezliği ve adrenal kalsifikasyon mevcutmuş. Olguların 6‟sının 12. aya kadar yaşadığı gözlenmiş; daha önce benzer klinik özellikleri gösteren 21 olgunun retrospektif incelemesinde; tamamının 12. aya ulaşmadan kaybedildiği belirtilmiştir. Kasım 2014 itibariyle bu çalışma dahilinde olan 6 olgunun ortalama yaşı 22 aylık olarak saptanmış; halen 5 olgunun enzim replasman tedavisi devam etmektedir. Kaybedilen 4 olgu sebelipase alfa tedavisini 4-5 kez aldıktan sonra kaybedilmiş olup; ölüm sebepleri sebelipase alfa ile ilişkili bulunmamış; hastalığın komplikasyonlarına bağlanmıştır. Ayrıca çalışmada sebelipase alfa tedavisiyle olguların hepatosplenomegalisinde azalma, kilo alımında artış, gastrointestinal semptomlarında azalma ve biyokimyasal-hematolojik parametrelerinde düzelme gözlenmiştir (52).
Rekombinant insan lizozomal asid lipazı olan sebelipase alfa; bu çalışmalar doğrultusunda KEDH ve WH için Ağustos 2015‟te onay almış olup; bu hastalıkların tedavisinde kullanılmaktadır.
2.2. Ön Göstergeler
2.2.1. Kitotriozidaz
Kitin yapısı ve kitinazlar
Kitin polimerleri böceklerin dış iskelet yapılarında, birçok mantar ve olgun hücre duvarında ve nematodlarda yapısal element olarak bulunan ve N-asetil glukozamin rezidülerinin β,1-4 bağlanması ile oluşmuş bir polisakkarit olup;
18 dünyada selülozdan sonra en fazla bulunan polisakkariddir (53). Bu patojenler kitini konakçı oldukları hücrede kendilerini zor şartlardan korumak için kullanırlar. Kitin yokluğu patojenin ölümüne neden olabilir.
Kitin depolanması; biyosentez ve yıkımı ile düzenlenir (54). Birçok organizmada (bakteri, fungus, böcekler, bitkiler, virüsler ve protozoan parazitlerde)kitindeki β-1,4 bağlarını hidroliz eden kitinaz enzimleri bulunmaktadır (53). Bu enzimler daha çok düşük canlı türlerinde incelenmiştir. Kitinaz enzimleri, kitin içeren organizmalar ile oluşan infeksiyonlara karşı geliştirilen savunma sırasında çok miktarda üretilirler. Bu olay, kitin içeren patojenlere karşı gelişen doğal immun cevabın bir parçası olarak düşünülebilir. Ayrıca, kitin içeren mantar ve parazitlerde, kitinaz üretimi, hayat döngülerinin bir parçası olarak rol alır ve konak hücredeki yaşam sikluslarını tamamlamak için kullanılır. Sonuç olarak, patojenlerden korunmada kitinin önemi ve birçok patojenin yaşam siklusunda uygun miktarlarda kitinaz üretiminin öneminden dolayı, kitin sentez inhibitörleri ve kitinaz inhibitörleri, böcekleri, fungal patojenleri ve helmintik parazitleri ortadan kaldırmak için mikrobiyal biyokontrol ajanların ve yeni biyopestisitlerin geliştirilmesi için farmakolojik çalışmalarda yoğun ilgi odağı olmuştur (54).
Kitinazlar, yapısal ve reaksiyon mekanizmalarındaki farklılığa bağlı olarak 18 ve 19 glikozid hidrolazlar olarak 2 gruba ayrılırlar (53). Ayrıca, endokitinaz ve ekzokitinaz olarak da sınıflandırılabilirler. Endokitinazlar, kitin polimerinin iç kısmındaki beta glikozidik bağları gelişigüzel hidrolize ederek, baslıca düşük molekül ağırlıklı kitopolimerleri (örneğin kitotrioz, kitotetraoz gibi) oluştururlar. Ekzokitinazlar ise, N-asparajin bağlı glikoproteinlerin yıkımı ve kitin polimerinin indirgen ucundan kitobioz koparabilen kitobiyazlardır. Ayrıca, glikozid hidrolaz 18 (GH18) protein ailesine dahil olmayan, kitinin indirgen olmayan ucundan N-asetil β-D-glukozamin koparan β-hekzominidaz enzimleri de kitinaz grubu enzimlerdir (55).
İnsan vücudunda yapısal eleman olarak kitin bulunmaz ve kitini sentez veya metabolize ettiğine dair herhangi bir bilgimiz yoktur. Doğal olarak kitinaz enziminin de bulunmaması beklenirse de ilginç olarak insan genomunda kitinaz ve benzeri proteinleri kodlayan GH18 ailesine ait 8 adet gen bulunmuştur (55). İlk kez, 1994‟de Hollak ve arkadaşları; semptomatik Gaucher hastalarının plazmalarında aşırı artmış (100-1000 kat) kitinaz aktivitesi saptamışlar ve bu kitinaza, sentetik substrat
19 kitotrioz‟a da etki etmesi nedeniyle “kitotriozidaz” adını vermişlerdir (56). Deneysel veriler, kitotriozidazın hangi sınıf kitinaz (endo veya ekzokitinaz) olduğuna dair açık bir bulgu göstermemekle beraber, X-ray kristal çalışmasına dayanarak, bu enzimin endokitinaz olduğu ileri sürülmüştür (57).
İnsanlardaki GH18 ailesindeki 8 proteinden 3 tanesinin enzim aktivitesi (kitinaz) vardır. Bunlar a-) aktiflenmiş makrofajlarda depolanan ve sekrete edilen ve fungostatik etkisi olduğu belirlenen kitotriozidaz, b-) daha çok gastrointestinal kanal ve akciğerlerde eksprese olan ve doğal immünite ve besinlerin sindiriminde rol aldığı ileri sürülen asidik memeli kitinaz (AMCase) c-) kitin yanında asparajinle bağlanmış glikoproteinlerin lizozomal yıkımında rol alan di-N-asetilkitobiaz (58).
Kitotriozidazın gen yapısı: İnsan kitotriozidaz (OMIM 600031) cDNA‟sı 1995‟de Boot ve arkadaşları tarafından klonlanmış ve aminoasit dizisi belirlenmiştir (59). İnsan kitotriozidaz geni 1q31-q32 de bulunur ve 12 ekzondan olusan 20 kb‟lık dizi içerir (60)(Şekil 5).
ġekil 5: Kitotriozidazın yapısı (62)
İnsanlarda kitinaz protein ailesinin (GH 18 gen ailesi) bilinen diğer tüm üyelerinin genleri de kitotriozidaz geni gibi 1. kromozomda bulunmaktadır. İnsan kitotriozidaz gen dizisi, asit memeli kitinaz geni ile %57 oranında benzerlik gösterir (61).
Kitotriozidazın Doku Ġfadesi
Kitotriozidaz aktivitesi karaciğer, böbrekler, dalak, akciğerler ve kemik iliğinde saptanmışsa da baslıca makrofajlar tarafından çesitli uyaranlara (prolaktin, TNF-α, gama-interferon, lipopolisakkarit gibi) cevaben aşırı kitotriozidaz sentez ve salınımı olmaktadır (62). Kitotriozidaz monositlerde yoktur fakat matür insan
20 makrofajlarında eksprese olur. Hücre kültürlerinde, kitotriozidaz mRNA sadece monositlerin aktive makrofajlara dönüşümünün son aşamasında eksprese olur. Kitotriozidaz, matür insan makrofajlarından 50 kDa‟luk aktif enzim halinde sekrete edilirken, bir kısım enzim makrofajın lizozomlarında proteolitik işleme uğrar ve sonuçta kitinaz aktivitesine sahip 39 kDa‟luk forma dönüşerek depolanır (63). Sağlıklı bireylerin kanında kitotriozidaz‟ın ana kaynağı, lenfositler ve monositler değil, insan polimorf nüveli nötrofil (PMN)‟leridir (63). Enzim, bu hücrelerde 50 kDa‟luk Kitotriozidaz formu halinde sentezlenir ve özel granüllerde depolanır.
Kitotirozidazın iĢlevleri
Kitotriozidaz enziminin özellikleri detaylı olarak saptanmış olsa da fonksiyonları tam olarak aydınlatılmamıştır. Funguslara karşı defansta rolü olabileceğine dair kanıtlar vardır. Fungal ajanlara karşı primer defansda rolü olan makrofajlar, kitotriozidazın ana kaynağıdır (63).
Kitotriozidaz yapısal bir enzim değildir. Makrofajlar özel koşullar altında bu enzimi büyük miktarlarda (bazı koşullarda sekrete edilen proteinin yaklasık %1‟ini oluşturur) üretebilmektedir (62). Makrofajlar genellikle birçok organda doğal dirençte rol alan önemli elemanlar olarak düşünülmektedir. Makrofajların interferon-gama (IFN-γ), tümör nekrozis faktör alfa (TNF α) ve lipopolisakkarid (LPS) ile uyarılması sonucunda kitotriozidaz aktivitesi ve kitotriozidaz 1 mRNA düzeylerinde yükseklik saptanmıştır. Bu da kitotriozidazın çesitli lizozomal ve hematolojik hastalıklarda makrofaj aktivasyonunun biyokimyasal belirteci olmasının yanı sıra immunolojik cevabın bir bileşeni de olabileceğini düşündürmektedir (62). Kitotriozidaz enzimi inflamatuar bir protein olarak düşünülebilir zira aktive makrofajlardan sekrete edilir. Fakat üretimi, hücre kültüründe uyarımdan en az 1 hafta sonra olur ve zamanla artar. Bu nedenle, bir akut reaktif proteinden çok kronik bir inflamatuar belirteç olarak kabul edilebilir (65).
Kitotirozidaz’ın lizozomal depo hastalıklarında klinik kullanımı
Serum/plazma kitotriozidaz aktivitesi ölçümü, günümüzde lizozomal depo hastalıklarının tanı, tedavi ve takibi, sarkoidoz tanı ve tedavisinin izlenmesi gibi klinik uygulamalarının yanı sıra, çesitli araştırma çalışmalarında (başta paraziter infeksiyonlar ve bağışıklık mekanizması ile ilgili araştırmalar olmak üzere,
21 ateroskleroz, bazı hematolojik hastalıklar, nörolojik hastalıklar gibi) yaygın olarak kullanılmaktadır.
Serum kitotriozidaz aktivite ölçümünün en yaygın kullanım alanı, Gaucher hastalığının tanısı ve tedavisinin takibinde biyokimyasal gösterge olarak kullanılmasıdır. Hollak ve arkadaşları ilk kez 1994‟de Gaucher hastalarının serumlarında normal değerlere göre 100 ile 1000 kat arasında değişen bir yükseklik göstermişler ve tanıda dolaylı belirteç olarak kullanılmasını önermişlerdir (56). Günümüzde metabolizma merkezlerinde serum kitotriozidaz ölçümü bu amaçla rutin olarak kullanılmaktadır.
Plazma kitotriozidaz seviyesinde daha ılımlı yükseklikler diğer lizozomal depo hastalıklarından Niemann-Pick, GM1-gangliosidozis ve Krabbe hastalığında gösterilmiştir (60).
Fukozidozis, galaktosialidoz, glikojen depo hastalığı IV, Alagille sendromu, vekonjenital herpes virus infeksiyonuna bağlı hidrops fetalisde de yükselmiş kitotriozidaz aktivitesi bulunmuştur (66). Mukopolisakkaridozların az bir kısmında belirgin yüksek plazma kitotriozidaz aktivitesi görülmüş. Diğer lizozomal depo hastalıklarının çoğunda (ki birçoğunda intrasellüler lipid depolanması yoktur) yüksek plazma kitotriozidaz aktivitesi görülmemiştir (67).
Son zamanlarda Boot ve arkadaşları aterosklerotik dokuda kitotriozidaz aktivitesinin 55 kat arttığını göstermişlerdir (68). Ayrıca; insan aterosklerotik damar duvarı içindeki lipid yüklü makrofajlar ile kitotriozidaz ekspresyonu arasında açık bir bağlantı saptanmıştır. Artieda ve arkadaşları da aterosklerotik hastaların serumlarında kitotriozidaz aktivitesini ölçmüşler ve anlamlı derecede yüksek bulmuşlardır (65). Aterosklerotik lezyonun şiddetiyle de bağlantılı olması ateroskleroz boyutunun belirteci olabileceğini düşündürmektedir.
KEDH tanılı 135 olgunun ele alındığı bir derlemede; kitotriozidaz aktivitesinin diğer lizozomal depo hastalıklarında olduğu gibi, kolesterol ester depo hastalığınında tanı ve takibinde kullanılabiliceği belirtilmiştir (1).
22
2.2.2. Sitokeratinler ve CK-18
Cytokeratin 18 (CK18), karaciğerde bulunan majör fibriler bir proteindir ve hepatik apopitozis sırasında ortaya çıkan kaspazların (aspartat spesifik cystein proteazlar) en bilinen substratıdır (69). Apopitozis esnasında CK 18, kaspazlarla aspartat 238 ve aspartat 396 noktasında kırılır. M30 monoklonal antikor, CK-18‟in aspartat 396‟da kırılan fragmanını (M30 antijen) tanır. Böylece CK‟ler apopitotik marker olarak kullanılabilir (70)(Şekil 6).
Sağlıklı karaciğerde mitozla çoğalan benzer çok sayıda hücre apopitozla vücuttan uzaklaştırılır ve böylece organ homeostazı sağlanır . Farklı patofizyolojik durumlarda hücre proliferasyonu ile hücre ölümü arasındaki bu denge bozulabilir ve apopitotik yollar aktive olabilir. Yeni araştırmalarda kronik karaciğer hastalığının patogenezinde hepatik apopitozisin temel bir rol oynadığı gösterilmiştir (71). Hepatositlerdeki apopitotik hücre ölümü sonrasında kan dolaşımına kaspazlar tarafından kırılan CK18 salınımı olmaktadır .
ġekil 6: Kırılmış CK-18 (71)
Apopitotik yolun aktive olduğu karaciğer hastalıklarında CK18 düzeyleri artabilir. Bu hastalıkları şöyle sıralayabiliriz: Hepatosellüler karsinom, viral hepatitler, alkolik hepatit, non-alkolik steatohepatitis, kolestatik karaciğer hastalıkları, hepatik fibrozis ve siroz (72). CK18, karaciğer dışında pankreas, barsaklar ve epitelyal dokularda bulunur. CK18‟ in epitelyal kanserlerin tümörgenezisinde önemli rolü olduğu belirtilmektedir (73). Birçok epitelyal kökenli karsinomlarda (hepatosellüler karsinom da dahil) CK18 potansiyel tümör markerı olarak çalışılmıştır. Hepatosellüler karsinomun rekurrens ve metastazlarında da CK18 yüksek bulunmuştur. Bu nedenle kronik karaciğer hastalığının tanısında ve
23 evrelendirilmesinde CK18‟in non-invaziv bir biyomarker olarak kullanılabileceği düşünülmektedir (74).
Bantel ve arkadaşları, kronik HCV enfeksiyonunda erken karaciğer hasarını saptamada, serum CK18 düzeylerinin aminotransferazlara göre daha duyarlı olduğunu göstermişlerdir (75). Kronik HCV‟de CK18 ile AST, USG‟de steatoz ve histolojik steatoz skoru arasında pozitif korelasyon bulunmuştur (76). Yine HCV ile yapılan yeni bir çalışmada CK18, minimal/hafif histolojik lezyonlarda, orta/ağır histolojik lezyonlara göre anlamlı olarak daha düşük bulunmuştur (77). HBeAg (-) kronik hepatit B‟de CK18 düzeyi, inaktif HBV taşıyıcılarına göre anlamlı olarak daha yüksek bulunmuştur (78). CK18 ile ilgili en çok araştırma yapılan karaciğer hastalığı nonalkolik yağlı karaciğer hastalığı (NAFLD) olmuştur. Bu hastalıkta CK18‟in, karaciğer fibrozisinin varlığı ile korele olduğu ve hepatik inflamasyonda indikatör olabileceği gösterilmiştir . Ayrıca nonalkolik steatohepatitis-hepatik steatoz ayrımında, ALT normal NAFLD tanı doğrulamasında önemli bir test olabileceği gösterilmiştir (79).
2.2.3. C-Reaktif Protein Ve Yüksek Duyarlıklı CRP ( hsCRP)
C-reaktif protein (CRP) inflamasyon markerlarından biridir. İlk kez 1930 yılında Tillet ve Francis tarafından akut infeksiyonu olan hastalarda Streptococcus pneumonia‟nın C polisakkaridine bağlanan akut faz proteini olarak tanımlanmıştır. CRP diğer akut faz proteinleri gibi mikrobiyal enfeksiyonlar, doku yaralanması, otoimmün hastalıklara yanıt olarak karaciğerden sentezlenir. İnterlökin-1beta (IL- 1beta) ve IL-6 hepatositlerden CRP‟nin sentezlenmesini güçlü bir şekilde uyarır (80,81). Önceleri yalnız karaciğerden sentezlendiği düşünülürken, daha sonraları monositler, lenfositler, adipositler, aterosklerotik plaklar, koroner arter düz kas hücreleri, aort endotel hücrelerinde, Alzheimer hastalarının nöronlarında, inflamatuar uyarılardan sonra renal kortikal tübüler epitelinde de üretildiği bulunmuştur (81). CRP‟nin asıl etkisi patojenlere bağlanarak fagositik hücreler tarafından tanınması ve ortadan kaldırılmasını sağlamaktır. CRP aynı zamanda kompleman sistemini de aktive eder. Böylece inflamasyonun hem humoral hem de selüler basamağında rol almış olur (82). CRP düzeyi akut inflamatuvar durumlarda veya doku hasarında geçici olarak artmakta, kronik inflamatuvar olaylarda ise sürekli yüksek değerler izlenmektedir (83). CRP düzeyleri akut myokard infarktüsü, stres, travma,