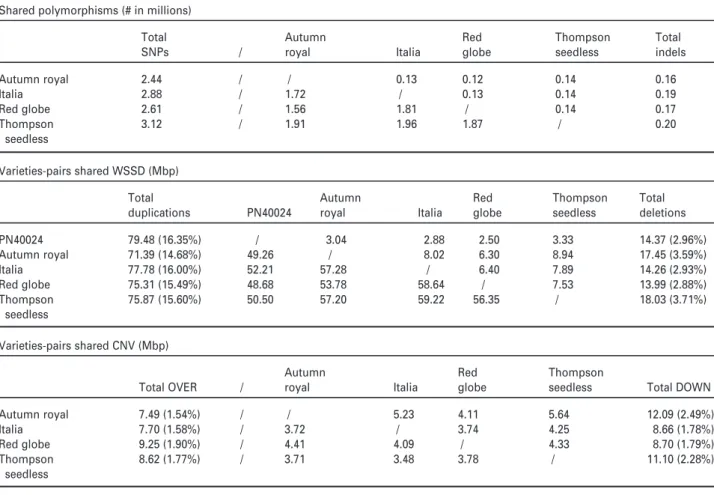
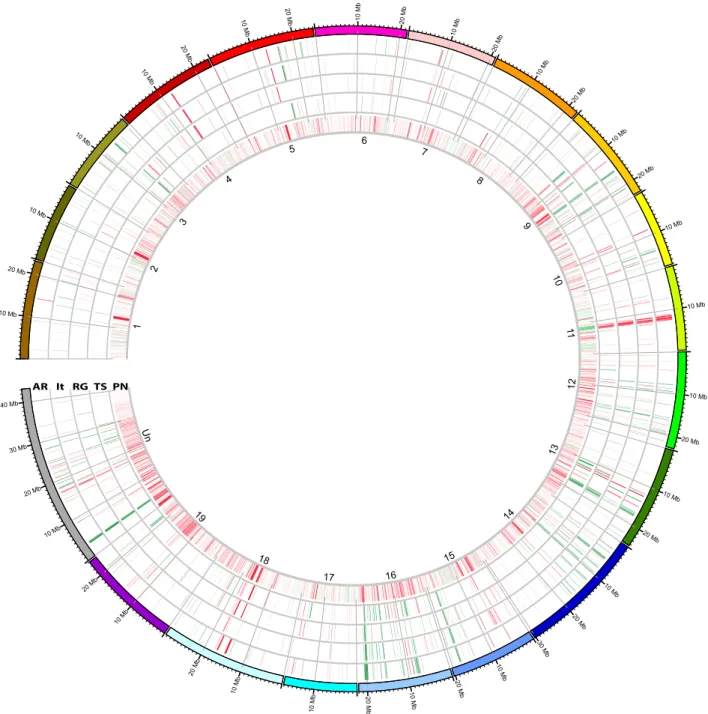
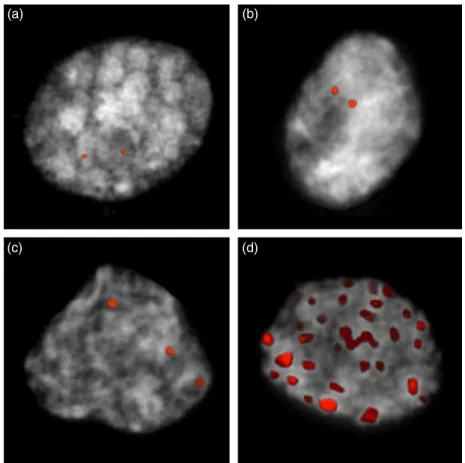
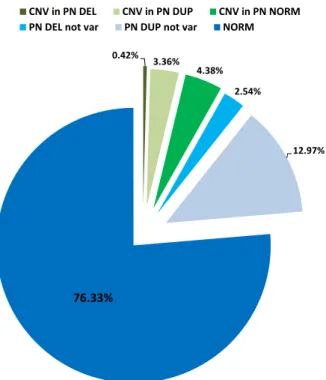
Inter-varietal structural variation in grapevine genomes
Tam metin
Şekil
Benzer Belgeler
Osmaniye Kanunnamesi, İslam ülkelerinde yapılan ilk yasal yurttaşlık düzenlemesi olması bakımından önemlidir. Ayrıca Kanunname o za- mana kadar olan Osmanlı
Amaç: Çalışmamızda diyabetik ayak ülserleri (DAÜ) gelişen hastalarda izole edilen mikrobiyal ajanları ve bu ajanların antibiyotik duyarlılık profillerini
Bu sonuçlara göre Çözelti C serisinden büyütülen CoCu filmlerde de Co filmlerde ve B çözeltisinden büyütülen CoCu filmlerde oldu ğ u gibi demanyetizasyonun bir
Here, we study the nonequilibrium Hall response following a quench where the mass term of a single Dirac cone changes sign and apply these results to understanding quenches in
Ayrıca kopolimer içerisindeki monomer bileşim oranları, akrilamitin monomerinin azot atomu içermesi, maleik anhitritin ise içermemesinden yararlanılarak, element
If each attribute is first measured on its own natural scale, the alternatives are located on each scale, and the minimum (maximum) location values are extrapolated to a common
Geçerlik güvenilirliğe yönelik yapılan istatistiksel değer- lendirmeler sonucunda hemşirelik lisans öğrencilerinin klinik uygulamalar sırasında algıladıkları stres türü
The following were emphasized as requirements when teaching robotics to children: (1) it is possible to have children collaborate in the process of robotics design, (2) attention
