T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLİM DALI
NİLOTİNİB İLE İNDÜKLENMİŞ KML HÜCRE MODELİ
APOPTOZUNDA JAK-STAT YOLAĞI
ELEMANLARINDAN STAT5A VE STAT5B’NİN
ETKİLERİNİN ARAŞTIRILMASI
Uzmanlık Tezi
Dr. Tunzala Yavuz
Tez Danışmanı
Prof. Dr. Güray Saydam
ii
ÖNSÖZ
İç Hastalıkları uzmanlık eğitimim boyunca bana her zaman ve her konuda destek
olan, hoşgörüsüyle hep yanımızda olduğunu hissettiren İç Hastalıkları Anabilim Dalı Başkanı Sayın Prof. Dr. Fehmi Akçiçek’e,
Bilimsel gelişimime büyük katkıları olan, bilgi ve tecrübelerinden faydalandığım, bana emeği geçen tüm İç Hastalıkları Anabilim Dalı Öğretim üyelerine,
Bu çalışmanın planlanması ve yürütülmesinde duyarlı ve anlayışlı yaklaşımı ile yanımda olan ve desteğini hiç eksik etmeyen sevgili danışman hocam Sayın Prof. Dr. Güray Saydam’a,
Tez çalışmamın temelini oluşturan ileri laboratuvar teknikleri konusunda, benimle her türlü bilgi ve birikimlerini paylaşan, her aşamada yardımcı ve destek olan Tıbbi Biyoloji Anabilim Dalı’ndan Araş. Gör. Burçin Tezcanlı Kaymaz’a,
Eğitimim ve gelişimime verdikleri büyük önem sayesinde buraya kadar gelebildiğim, sınırsız sevgileri ile bana güç veren babam Vidadi Guliyev’e, annem Safura Guliyeva’ya, bana her zaman yardım eden kardeşlerime,
Uzmanlık eğitimim boyunca sevgi ve sabırla beni her zaman destekleyen, hep yanımda olan sevgili eşim Ali Yavuz’a ve kayınvalidem Birgül Yavuz’a,
Tıpta uzmanlık sınavından 3 hafta önce varlığını bildiren, sınavda heyecanımı benimle birlikte yaşayan, beni sakinleştiren, eğitim sürecinde dünyaya gelip bu süreçte büyüyen, yoğun nöbetli dönemlerimden en çok etkilenen, bana her zaman yaşama sevinci ve geleceğe umut veren, canım oğlum Berk Can Yavuz’a, sonsuz teşekkürlerimi sunuyorum.
iii
İ
ÇİNDEKİLER
Simgeler ve Kısaltmalar Dizini... v
Şekiller Dizini ...vii
Tablolar Dizini ...viii
Grafikler Dizini ... ix
Özet ... x
Abstract ...xii
1. GİRİŞ VE AMAÇ ... 1
2. GENEL BİLGİLER ... 4
2.1. Tanım , Epidemiyoloji, Etiyoloji ve Tarihçesi ... 4
2.2. Patogenez. ... 6
2.2.1 KML Moleküler Genetiği ... 6
2.2.1.1 Ph kromozomu ... 6
2.2.1.2 ABL geni ve proteini ... 6
2.2.1.3. BCR Geni ve Proteini ... 7
2.2.1.4. Bcr-Abl Füzyon Geni ve Füzyon Proteini ... 8
2.2.2. KML’de Moleküler Patogenez ... 9
2.2.2.1BCR-ABL1 Aracılı Lösemik Transformasyonun Moleküler Mekanizması ... 9
2.2.2.2 Lösemik Dönüşümde Rol Oynayan Sinyal İleti Mekanizmaları ... 11
- MAP Kinaz ve Ras/Raf/MEK/ERK Sinyal İletim Yolu ... 12
- PI-3 Kinaz/Protein Kinaz B Sinyal İletim Yolu ... 13
- Myc Sinyal İletim Yolu... 13
- JAK-STAT Sinyal İletim Yolu ... 13
2.2.2.3. STAT5’in genel özellikleri ... 17
2.3. KML’de Tanı ... 18
2.4. KML’de Ayırıcı Tanı ... 19
2.5. KML Klinik evreleme ve prognoz ... 20
2.6. KML Tedavisi ... 22
2.6.1.Tirozin kinaz inhibitörleri ... 25
2.6.2 Allojeneik Hematopoetik Kök Hücre Nakli... 30
iv
3. MATERYAL VE METOD ... 34
3.1. Lösemik Hücre Hattı Ve Hücre Kültürü ... 34
3.1.1. K562 KML hücre hattının özellikleri... 34
3.1.2. K562 hücre hattında kullanılan besiyeri ve kültür işlemleri ... 34
3.1.3. Dondurulmuş hücre hattının çözülmesi ... 35
3.1.4. Hücre hattının pasajlanması ... 35
3.1.5. Hücre sayımı ve canlılığın değerlendirilmesi ... 36
3.2.Sitotoksisite Çalışmaları... 32
3.3. PCR ile STAT 5A ve 5B transkript düzeylerinin saptanması... 39
3.3.1.K562 hücrelerinde STAT Ekspresyonlarının Belirlenmesi ... 39
3.3.2. Total RNA İzolasyonu ... 39
3.3.3.cDNA Reaksiyonu ... 40
3.3.4. STAT Ekspresyonlarının Belirlenmesi ... 40
3.3.5. STAT5A, STAT5B mRNA Ekspresyon Değerlerinin Belirlenmesi ... 41
3.4. STAT 5A ve 5B ekspresyonlarının protein düzeyinde saptanması ... 43
3.4.1.I-Blot Dry Blotting Sistem ile Western Blotlama ... 43
3.4.2.WesternBrezze Chromogenic İmmunodetection Protokol Uygulaması .... 44
3.4.3. Protein Ekstraksiyonu ... 45
3.4.4. Bradford Metoduna Göre Protein Tayini ... 46
3.5.Apoptozisin saptanması ... 46
3.5.1. Cell Death Detection Kit... 46
3.5.2. Caspase-3 aktivity deneyi ... 49
3.6.İstatistiksel Değerlendirme... 50
4. SONUÇLAR ... 51
5. TARTIŞMA ... 58
v
SİMGELER VE KISALTMALAR DİZİNİ
ABL : Abelson geni
AHKHN : Allojeneik Hematopoetik Kök Hücre Nakli AKT (protein kinaz B) : Serin-treonin spesific protein kinase
ALL : Akut lenfoblastik lösemi AML : Akut myeloid lösemi
A-MuLV : Abelson Mürin Lösemi Virüsünün Arg : Abl ile ilgili gen
ATP : Adenosin trifosfat BAP-1 : Bcr-associated protein-1 BCR : Breakpoint cluster region BİM : Bcl-1 Interacting medyatörü
DASISION : Dasatinib versus İmatinib Study in Treatment Naive CML-CP Patients
DSÖ : Dünya Sağlık Örgütü ELN : European Leukemia Net
ENESTnd : Evaluating Nilotinib Efficacy and Safety in Clinical Trials of Newly. Diagnosed Ph+ CML Patients
EPO : Eritropoetin
ERK : Extracelluler signal regulated kinase FDA : Food and Drug Administration FISH : Fluoresans in situ hibridizasyon G- CSF : Granulosit koloni stimule edici faktör G6PDH : Glukoz 6 fosfat dehidrogenaz
GAP : GTPaz-aktive edici protein GDP : Guanidin difosfat
GEF : Guanidin exchange faktör GH : Growth hormon
GİST : Gastrointestinal Stromal Tümör
GM- CSF : Granulosit- makrofaj koloni stimule edici faktör GMÖ : Granülosit/makrofaj öncülleri
GTP : Guanidin trifosfat
vi
HKH : Hematopoetik kök hücreleri IFN : İnterferon
IL : İnterlökin
JAK : Janus Kinaz
KML : Kronik myeloid lösemi LÖ : Lenfoid öncülleri
MAPK : Mitojen actived protein kinase M-bcr : Major breakpoint cluster region m-bcr : Minör breakpoint cluster region MEÖ : Megakaryosit/eritrosit öncülleri MMY : Major moleküler yanıt
MRNA : Messenger RNA
MTOR : Mammalian target of rapamycin MY : Moleküler yanıt
Myc : Myelositomatozis MÖ : Myeloid öncülleri
PDGF : Platelet derived growth factor – trombosit kaynaklı büyüme faktörü
Ph : Philadelphia kromozomu PI3K : Fosfatidil Inositol 3 Kinaz
PIAS : Protein inhibitors of activated STATs- aktive STAT’ların protein inhibitörleri
RT- PCR : Ters transkriptaz polimerize zincir reaksiyonu RT-Q-PCR : Real Time Quantitative Polymerase Chain Reaction SAPK : Stres activated protein kinase
SFK : SRC family of protein tyrosine kinase
SOCS : Suppresors of cytokin signaling- sitokin sinyalleşme supresörleri
STAT : Signal Transducers and Activators of Transcription SY : Sitogenetik yanıt
THY : Tam hematolojik yanıt TKI : Tirozin Kinaz İnhibitörü
vii
Ş
EKİLLER DİZİNİ
Şekil 1. Kronik Myeloid Löseminin gelişimi. Şekil 2. Philadelphia kromozomu
Şekil 3. Abl proteininin yapısı Şekil 4. BCR proteininin yapısı.
Şekil 5. Bcr ve Abl genlerindeki kırılma noktaları ve Bcr-Abl füzyon proteinleri Şekil 6. BCR-ABL geni tarafından kontrol edilen malign transformasyon
mekanizmaları
Şekil 7. BCR-ABL füzyon geninin etkili oldugu yolaklar Şekil 8. JAK-STAT Sinyal İletim Yolağı:
Şekil 9. STAT moleküllerinin yapıları
Şekil 10. IL-6 ile aktive olan JAK-STAT yolağı Şekil 11. KML, periferik yayma
Şekil 12. İmatinib mesilat Şekil 13. İmatinibin Kimyasal Yapısı
Şekil 14. Nilotinib ile ABL kinaz Şekil 15. Nilotinibin Kimyasal Yapısı
Şekil 16. Tripan Mavisi Boyası İle Hücrelerin Sayımı Şekil 17. Hücre proliferasyon kiti WST-1
Şekil 18. Cell Death Detection ELISA Kit ( Roche) Şekil 19. Caspase 3 Activity Assay Kit , (Roche)
Şekil 20. STAT5A ve STAT5B hedef genlerinin mRNA kopya sayılarını belirlemede
kullanılacak olan G6PDH referans geninin standart eğrisi
Şekil 21. STAT5A ve STAT5B protein düzeylerinin WesternBreeze® Chromogenic
viii
TABLOLAR DİZİNİ
Tablo 1.DSÖ KML akselere faz tanımı Tablo 2 .DSÖ KML blastik faz tanımı Tablo 3. KML Risk Skorlamaları
Tablo 4: ELN 2013 Kılavuzuna göre Kronik Myeloid Lösemi Tedavisi
Tablo 5. THD 2013 kılavuzuna göre kronik evre KML tedavisinde birinci, ikinci ve daha sonraki basamaklardaki tedavi önerileri
Tablo 6. THD 2013 Kılavuzuna göre Hızlanmış Evre ve Blastik Evre KML hastalarında tedavi önerileri
Tablo 7. İkinci kuşak TKİ seçimi
Tablo 8. İkinci kuşak TKI’lerinde mutasyonlara duyarlılık
Tablo 9. Birinci kuşak TKI (herhangi bir TKI) tedavisine yanıt tanımları Tablo 10. İmatinibe yanıtsız durumunda ikinci kuşak tedaviye yanıt tanımları Tablo 11. PVDF membranlar için çözelti hazırlama
Tablo 12. STAT5A gen ekspresyonunun zamana bağlı rölatif oranları Tablo 13. STAT5B gen ekspresyonunun zamana bağlı rölatif oranları
ix
GRAFİKLER DİZİNİ
Grafik 1. K562 KML hücre hattı için nilotinibin IC50 dozu Grafik 2. STAT5A gen ekspresyonunun zamana bağlı değişimi. Grafik 3. STAT5B gen ekspresyonunun zamana bağlı değişimi
Grafik 4. IC50 dozunda Nilotinib uygulanmış ve uygulanmamış KML K562 hücrelerinin Cell Death Detection ELISA Kit ile saptanan apoptoz analizleri Grafik 5. IC50 dozunda Nilotinib uygulanmış ve uygulanmamış KML K562
x
ÖZET
Nilotinib ile İndüklenmiş KML Hücre Modeli Apoptozunda
JAK-STAT Yolağı Elemanlarından JAK-STAT5A ve JAK-STAT5B’nin Etkilerinin
Araştırılması
Dr. Tunzala Yavuz
Ege Üniversitesi Tıp Fakültesi İç Hastalıkları Anabilim Dalı Bornova/ İZMİR
AMAÇ: Kronik Myeloid Lösemi (KML) multipotent kök hücrelerinin neoplastik
transformasyonu sonucunda ortaya çıkan klonal myeloproliferatif hastalıktır. t(9,22) translokasyonu sonucunda oluşan BCR-ABL füzyon geni KML’nin patogenezinde merkezi rol oynamaktadır. BCR-ABL füzyon geni kontrolsüz tirozin kinaz aktivitesi göstermekte ve bazı sinyal yolaklarını uyarmaktadır. JAK-STAT sinyal yolağı KML’nin patogenezinde rol oynayan yolaklardan en önemlisidir. JAK-STAT sinyal yolunda işlev gören STAT proteinleri fosforilasyon ile aktifleşirler. Bu sinyal yolağı, hücre büyümesi, proliferasyonu, farklılaşması, sağkalımı gibi birçok hücre süreçlerini etkiler. JAK-STAT yolağı elemanları STAT5A ve STAT5B; sinyal iletiminde ve malignitede ekspresyonu artan genlerin aktivasyonunda görevli olup, lösemi gelişiminde rol alırlar. STAT gen ekspresyonunun baskılanmasının tümör hücrelerinde apoptoz indüklenmesi ile sonuçlandığı gösterildikten sonra bu alanda çalışmalar hız kazanmıştır. Nilotinib, KML tedavisinde kullanılan tirozin kinaz inhibitörlerinden biridir.
Bu çalışmada; nilotinib ile induklenmiş KML hücre modeli apoptozunda JAK-STAT yolağı elemanlarından STAT5A, STAT5B-nin etkilerinin araştırılması ve apoptoz oranlarının belirlenmesi amaçlandı.
MATERYAL VE METOD: Nilotinibin sitotoksik efektif dozu IC50 değeri tripan
mavisi ve WST-1 hücre proliferasyon analizi testi ile belirlendi. Nilotinib muamelesi sonrasında hücrelerin apoptotik durumu “Cell Death Detection kit” ve “Caspase 3 Activity Assay kit” ile belirlendi. STAT5A ve STAT5B mRNA ekspresyon düzeyleri,
xi
nilotinib ile muamele sonrasında eş zamanlı olarak qRT-PCR ile belirlendi. STAT5A ve STAT5B protein düzeylerinin WesternBreeze® Chromogenic Kit–Anti-Rabbit” kit ile değerlendirilmesi yapıldı. İstatistik analizler Student T Test ile (Graph Pad Prism Programı) anlamlılık düzeyi p<0.05 kullanılarak değerlendirilmiştir.
BULGULAR: Nilotinib’ in IC50 dozu, 48. saat için 34.5 nM olarak belirlenmiştir.
Nilotinib muamelesi sonrasında hücrelerin apoptotik durumu “Cell Death Detection kit” ve “Caspase 3 Activity Assay kit” ile belirlendi. Buna göre, IC50 dozunda nilotinib uygulaması sonrasında, kontrol grubuna göre “Cell Death Detection kit” ile 1.89 katlık (52.8% oranında) apoptoz artışı (p=0.0012), “Caspase 3 Activity Assay kit” ile 2.1 katlık apoptoz artışı saptandı (p=0.045). Hedef genlerin mRNAekspresyonlarını kıyasladığımızda, STAT5A ekspresyonu 72. saatte 2.87 kat (%65.12 oranında baskılanmıştır, p= 0.0033 ), 96. saatte 11 kat azalırken (%90.9 oranında baskılanmıştır, p<0.0001), STAT5B ekspresyonu 72. saatte 5.2 kat (% 80.9 oranında baskılanmıştır, p= 0.0032 ), 96.saatte 15 kat azalmıştır(%93.3 oranında baskılanmıştır, p<0.0001). STAT5A protein düzeyleri özellikle 96.saatte, STAT5B protein düzeyleri zamana bağlı olarak 24. saatten itibaren kontrol grubuna göre belirgin olarak baskılandı. Sonuçlar mRNA ekspresyon sonuçları ile uyumlu saptandı.
SONUÇ: Çalışmamızda K562 KML hücre dizisinde nilotinibin apoptozisi anlamlı düzeyde indüklediği ve JAK-STAT yolağı üzerinden, STAT5A ve STAT5B ekspresyonlarını baskılayarak bu etkiyi gösterdiği, apoptozda STAT5A, STAT5B’nin önemi gösterildi, bu da KML ve JAK-STAT mekanizması ile gerçekleşen bir çok malignitelerin tedavisinin hedefi olarak, STAT5A ve STAT5B’ni hedef alan tedavi yöntemlerinin üzerinde çalışmalar yapılması gerektiğini düşündürmektedir.
xii
ABSTRACT
Investigating The Role of JAK-STAT Pathway’s elements STAT5A
and STAT5B upon Nilotinib Induced Apoptosis for CML Cell Model.
Dr. Tunzala Yavuz
Ege University Faculty of Medicine Department of Internal Medicine Izmir/TURKEY
AIM: Chronic Myeloid Leukemia (CML) is a clonal disorder which is based on
neoplastic transformation of a multipotent stem cell. BCR-ABL fusion gen occurs as a result of translocation t(9,22). BCR-ABL fusion gene play a central role in the pathogenesis of CML. BCR-ABL fusion gene has a limitless tyrosine kinase property and induces several signal transduction pathways. JAK-STAT signaling pathway is one of the most important of pathways in the pathogenesis of CML. STAT proteins function in the JAK-STAT signaling pathway and are activated by phosphorylation. As a result of this signaling event, they affect many cellular processes including cell growth, proliferation, differentiation, and survival. STAT5A and STAT5B ; members of JAK-STAT signaling pathway; are assigned in signal transduction and in the activation of the genes whose expressions increase in malignancy and also play an important role in the development of leukemia. After it has been found out that suppression of STAT gene expression results in induction of tumor cell apoptosis, investigations made in this area have been accelerated. For CML therapy, tyrosine kinase inhibitors are widely being used and one of them is nilotinib.
In the current study, we aimed to determine transcriptional and translational differences of STAT5A and STAT5B and also apoptosis rates following nilotinib treatment in CML model K562 cells.
METHODS: By using trypan blue and Cell Proliferation Reagent WST-1, we
calculated the cytotoxic effective dose IC50 concentration of nilotinib. The apoptotic case of the cells was assessed by ‘Caspase 3 Activity Assay Kit’ and ‘Cell Death Detecton Kit’ following the same treatments with same time intervals. While mRNA
xiii
expression levels of STAT5A and STAT5B were also determined by real time qRT-PCR following nilotinib treatment for the same time interval. Protein expression analyses were performed by Western Blot analysis according to ‘WesternBreeze Chromogenic Kit-Anti-Rabbit’ kit manuel instructions. Statistical analyses were done by GraphPad prism sofware with a significance of p<0.05
RESULTS: The IC50 dose of nilotinib was determined as 34.5 nM for 48th hour.
While number of apoptotic cells were increased by 2.1 fold, (p=0.045) as a result of measurement of caspase 3 activity; a 1.89 fold, 52.8% increase was detected in apoptosis rate following Cell Death assay (p=0.0012) in nilotinib treated group. As for mRNA expression results, while STAT5A expression was significantly decreased by 65.12% [(2.87 fold; p= 0.0033)] and by 90.9% [(10.99 fold; p<0.0001)] at 72th – 96th hours respectively; STAT5B was downregulated by 80.95% [(5.25 fold; p= 0.0032), and 93.36% (15.08 fold; p<0.0001)] for 72th and 96th hours in nilotinib treated group compared to control group. As for protein results, both STAT5A and STAT5B protein expression levels were inhibited in a time dependent manner but dramatic suppressions were detected especially at 96th hour for each protein.
CONCLUSION: According to the results of this study, nilotinib induced apoptosis
through STAT pathway via the supression of STAT5A and STAT5B expressions. Therefore, STAT5A and STAT5B are important moleculer targets in the research of CML pathogenesis.
Thus, targeting STAT5A and STAT5B may be an effective strategy for the treatment of CML and other myeloproliferative diseases.
1
1.GİRİŞ VE AMAÇ:
Kronik Myeloid Lösemi (KML) multipotent kök hücrelerinin neoplastik transformasyonu sonucunda ortaya çıkan klonal myeloproliferatif hastalıktır. KML özgül kromozom anomalisi ile tanımlanan ilk kanser tipidir. KML vakalarının %90’ından fazlasında Philadelphia (Ph) kromozomu görülmektedir. Ph kromozomu oluşurken ABL onkogeni 9 numaralı kromozom üzerinde bulunan 9q34.1 bölgesinden 22 numaralı kromozom üzerindeki 22q11.2 BCR bölgesine aktarılmaktadır [t(9;22)(q34;q11)]. Bu translokasyon sonucunda KML’nin patogenezinde merkezi rol oynadığı düşünülen BCR-ABL füzyon geni oluşur. Yeni gen, artmış tirozin kinaz aktivitesine sahip bir onkoprotein kodlar. BCR-ABL onkoproteini çeşitli hücre içi sinyal iletim yolaklarını aktive ederek anormal hücresel adhezyona, artmış hücresel proliferasyona ve apoptozisin baskılanmasına yol açar.
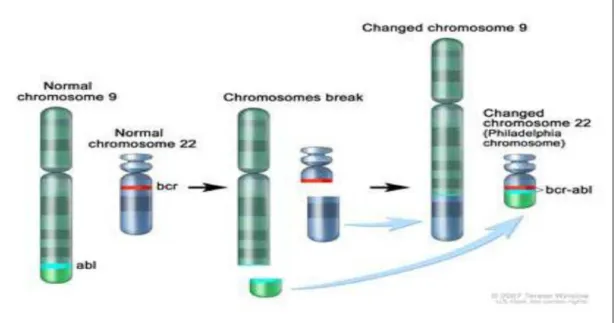
Hematopoetik kök hücrelerinin (HKH) BCR-ABL füzyon gen ekspresyonuyla hastalık başlar (Şekil 1.)
HKH’ ler myeloid öncülleri (MÖ) ve lenfoid öncülleri (LÖ) şeklinde farklılaşır. MÖ’ ler granülosit/makrofaj öncülleri (GMÖ) ve megakaryosit/eritrosit öncülleri (MEÖ) ; GMÖ’ler granülosit öncülleri (G) ve makrofajlar (M) şeklinde, MEÖ’ler ise eritrositler, megakaryositler ve onların ürünü trombositler şeklinde farklılaşır. Lenfosit öncülleri T hücreleri, B hücreleri şeklinde farklılaşır. Bu dönemde farklılaşma yeteneğini kaybetmemiş pluripotent kök hücrelerinden gelen Ph (+) lösemik klonunun çok fazla çoğalması söz konusudur. Bu hücrelerin farklılaşması sonuna kadar devam etmekte ve yüksek miktarda granülosit dolaşımda yer almaktadır. Kısaca KML’nin kronik fazı granülositik hücre serisinin kütlece artışı ile karakterizedir.
2 Şekil 1: Kronik Myeloid Löseminin gelişimi.
BCR-ABL aktivasyonu ile hücre içinde bir çok sinyal yolağı aktive olur. Bu yolaklardan JAK-STAT, RAS, PI3K , Src ailesi kinazların aktivasyonu ile apoptozis inhibe olmakta ve proliferasyon uyarılmaktadır. Sitokin aracılı STAT aktivasyonunda öncelikle sitokin, hücre yüzeyindeki reseptörüne bağlanır daha sonra reseptör ile ilişkili olan JAK proteinleri çapraz fosforilasyon ile aktive olur. Aktive JAK proteinleri reseptörü de fosforile ederler. Bu bölgeler sitoplazmadaki inaktif STAT proteinlerinin reseptör ile etkileşmesine olanak sağlar. STAT proteinleri daha sonra homodimer ya da heterodimer oluşturmak üzere reseptörden ayrılarak hücre çekirdeğine gelirler ve DNA üzerinde özgül cevap elemanı dizileri ile etkileşerek hedef genlerin transkripsiyonunu uyarırlar. STAT3 ve STAT5 aktivitelerinin kontrolsüz işleyişi malign transformasyonda rol oynamaktadır. STAT proteinleri iki mekanizma aracılığıyla karsinogenezde etkili olur. Bunlardan biri STAT’ın sürekli aktivasyonu diğeri ise proteinin c-ucunun mutasyona uğramasıdır. Devamlı olarak aktif olan STAT proteini antiapoptotik yolları uyararak malign süreçte etkili olabilir. İmatinib; erken evre KML ve birinci basamak tedavi olarak kullananların %75’inde sitogenetik yanıt sağlarken, ileri faz KML ve Ph(+) ALL’de tedaviye refrakterlik daha sıktır ve indüklediği remisyonlar erken relapslarla sonuçlanarak kısa ömürlü olmaya meyillidir. BCR-ABL’nin reaktivasyonuna
3
yol açan farklı mekanizmalar bu duruma neden olur. Bu durum imatinib direnci olarak karşımıza çıkmaktadır. İlaç direncine karşı koymak için yolağın kapsadığı diğer önemli sinyal bileşenleri yada BCR-ABL’yi diğer mekanizmalarla inhibe etmek hedeflenmektedir. Bu direnci yenmek için ikinci kuşak tirozin kinaz inhibitörleri geliştirilmiştir.
Bir aminoprimidin olan Nilotinib, ABL’ye yüksek oranda selektif olup, BCR-ABL kinaz inhibisyonu etkisi imatinib duyarlı KML hücreleri üzerinde imatinibden 20-30 kat, imatinib dirençli KML hücreleri üzerinde ise 3-7 kat daha potent olan ikinci kuşak tirozin kinaz inhibitörüdür. İmatinibe benzer sekilde BCR-ABL1’in ATP-bağlayıcı bölgesine kompetitif inhibisyonla bağlanarak etki eder, ancak bağlanma affinitesi ve ABL kinaz seçiciliği imatinibden üstündür.
Bizim çalışmamızda Nilotinib ile induklenmiş KML hücre modeli apoptozunda JAK-STAT yolağı elemanlarından STAT 5A ve STAT 5B’nin etkilerinin araştırılması amaçlandı.
4
2. GENEL BİLGİLER
KRONİK MYELOİD LÖSEMİ
2.1. Tanım:
KML, hematopoetik bir kök hücrenin neoplastik transformasyonundan kaynaklanan klonal bir myeloproliferatif hastalıktır [1]. KML; günümüzde moleküler düzeyde en iyi tanımlanmış lösemi tipi olup, 2008 Dünya Sağlık Örgütü (DSÖ) sınıflamasına göre; “Myeloproliferatif Neoplaziler ” arasında yer alır [2]. 9 ve 22. kromozomlar arasında karşılıklı translokasyon sonucu olusan Philadelphia kromozomu ile karakterizedir. Bu translokasyon sonucunda oluşan kimerik BCR-ABL geni kontrolsüz tirozin kinaz aktivitesi gösterir. Tirozin kinaz aktivitesi; hücre proliferasyonunu, maturasyonunu ve adhezyon sinyal yolaklarını aktive ederek, apoptozu baskılar ve malign hücre transformasyonuna yol açarak KML patogenezinin temelini oluşturur [3].
Epidemiyoloji:
KML, tüm erişkin lösemilerin %15-20’sini oluşturur. Yıllık insidansı yaklaşık 1 - 2/100.000’dir. Hastalık erkeklerde kadınlara oranla biraz daha sıktır. Yaşamın 5.ve 6. dekatlarında sık görülür ve yaşla birlikte insidansı artar. Olguların ancak % 10 kadarı 20 yaş ve altındadır [1]
Etiyoloji:
Genellikle etyolojide suçlanan izole tek bir ajan bulunmamaktadır ve olguların çoğu sporadik olgulardır. Bilinen ailesel predispozisyon olmayıp, alkilleyici ajanlar gibi sitotoksik ilaçlara maruz kalma ile net bir ilişkisi bulunamamıştır. Japonya’da 1945 yılındaki atom bombası patlamasından sonra KML insidansı artmıştır. Ancak nükleer endüstrilerde çalışanlarda, akilleyici ajan maruziyeti ya da radyoterapi sonrası KML riskinde artış gösterilememiştir. Bu veriler doza bağımlı bir etki olabileceğini düşündürmektedir [4].
5 KML’nin tarihçesi:
KML ilk olarak 1845 yılında Dr. Rudolf Virchow ve Dr. John Hughes Bennett adlarında iki patolog tarafından tanımlanmış [5, 6] . 19. Yüzyılın sonlarında kanın özel boyamalarla incelenmesi henüz gelişmemiş olduğundan, hastalıkla ilgili ilk açıklamalar sınırlı kalmış. Yaklaşık 100 yıl sonra; 1960 yılında Peter Nowell ve David Hungerford, KML hastalarında, kromozomal delesyon olarak düşünülen akrosentrik kromozom varlığını tanımlamışlardır. Bu durum tarihte bir kromozomal anormalliğin spesifik bir maligniteyle bağlantısının kurulmasının ilk örneğidir[7]. 1973 yılında Dr. Janet Rowley tarafından kromozom 9’daki ABL protoonkogenin 22.kromozomdaki BCR geni yakınına resiprokal translokasyonu gösterilmiştir. Bu yeni belirleyici, keşfedildiği
şehrin onuruna Philedelphia kromozomu (Ph) olarak adlandırılmıştır [8] (Şekil 2). Daha
sonra ABL ve BCR genleri tanımlanmış ve BCR-ABL füzyon proteininin tirozin kinaz aktivitesi olduğu ve onkogenezde anahtar rolü oynadığı gösterilmiştir. 2001 yılında Druker ve ark. tirozin kinaz aktivitesinin inhibisyonunun KML tedavisinde etkili olacağını düşünerek, bir tirozin kinaz inhibitörü olan STI571 (imatinib) geliştirmişlerdir. İmatinib, KML ve kanser tedavisinde bir dönüm noktasıdır [7-9]
6 2.2. PATOGENEZ:
2.2.1. KML Moleküler Genetiği: 2.2.1.1. Ph kromozomu:
Kronik Myeloid Lösemi (KML), özgül kromozom anomalisi ile tanımlanan ilk kanser tipidir. 1960 yılında Nowell ve Hungerford yedi KML’li hastanın hücrelerinde nokta akrosentrik kromozom tanımlamış ve Philadelphia ismini vermişler [7]. KML hastalarının %95’inde, akut lenfositik lösemili (ALL) çocukların %5’inde, erişkinlerin %15 -30’unda ve yeni tanı akut myeloid lösemili (AML) hastaların %2’sinde saptanmaktadır [10]. Ph kromozomu, t(9;22)-(q34;q11), 9. ve 22. kromozomların uzun kolları arasında resiprokal bir transformasyon sonucu oluşan bir kromozomdur. 9q34 deki ABL protoonkogeninin 22q11 deki BCR geni karşılıklı translokasyonu ile oluşan BCR-ABL füzyon geni ortaya çıkar [11]. (Şekil 2)
2.2.1.2. ABL geni ve proteini
ABL geni Abelson murine lösemi virusunda bulunan viral ABL geni ile benzer bir yapıda, 230 kb uzunluğunda bir gen olup 11 ekzon içerir. 9q34’ de lokalize olan ABL geni 145 kD’ luk bir reseptör olmayan tirozin kinazı kodlar [12] (Şekil 3).
7
ABL proteini nonreseptör protein kinaz olup hem sitoplazma hem de nükleus içerisinde bulunarak iki kompartman arasında geçişe izin verir[12, 13] .
Normal Abl proteini, çekirdekte, hücre siklusunun G1 fazında tutulmasını sağlayarak hücre büyümesinin negatif düzenleyicisi olarak rol alır. Ayrıca hücrelerin genotoksik ve oksidatif stres ortamına verdiği cevabı, p53 ya da bunun fonksiyonel homoloğu olan p73 aracılığıyla apoptozisi indükleyerek kontrol eder. Sitoplazmik Abl ise integrin ve PDGF (platelet-derived growth factor) sinyalizasyonunda rol alır [3, 13].
2.2.1.3. BCR Geni ve Proteini:
BCR (breakpoint cluster region) geni 22 nolu kromozom üzerinde q11.2 bant bölgesine lokalize olmuştur. 23 ekzondan oluşmaktadır. Gen ürünü olan protein 160kd
ağırlığında olup, birkaç işlevsel bölgeye sahiptir ve P160bcr olarak adlandırılır. BCR’nin
1. eksonu tarafından kodlanan N-terminalinden itibaren ilk 426 aminoasitlik bölge özellikle önemlidir çünkü transformasyon için gerekli olan homo-oligomerizasyonu düzenleyen ve serin/threonin kinaz aktivitesi gösteren “coiled-coil” bölgesini içerir. Serin/threonin kinaz; BCR’nin otofosforilasyonunu ve BAP-1’in (Bcr-associated protein-1) fosforilasyonunu sağlar [14, 15]. BCR proteininin 3-10. eksonları tarafından kodlanan 490-690. aminoasitler arasındaki merkezi sekansı, Dbl onkoprotein ve pleckstrin-homoloji bölgelerini içerir ve Rho-GEF bölgesi adını alır. Bu bölgeler Rho proteinleri için guanidin exchange faktörü olarak (GEF) fonksiyon görmekte ve G proteinlerinde guanidin trifosfat’ın (GTP) guanidin difosfat’a (GDP) dönüşümünü sağlayarak transkripsiyon faktörlerinin ve kinaz aktivasyonunun uyarılmasına neden olmaktadır [16]. Bcr’nin C-terminal ucu ise, Rac ve Cdc42 proteinleri için GTPaz-aktive edici protein (GAP) fonksiyonu olan bir bölgeye sahiptir. GAP bölgesi GTP hidrolizini uyararak, merkezi Rho-GEF bölgesinin aksine, tirozin kinaz aktivitesinin inhibisyonuna neden olur[17].(Şekil 4)
8 Şekil 4. BCR proteininin yapısı.
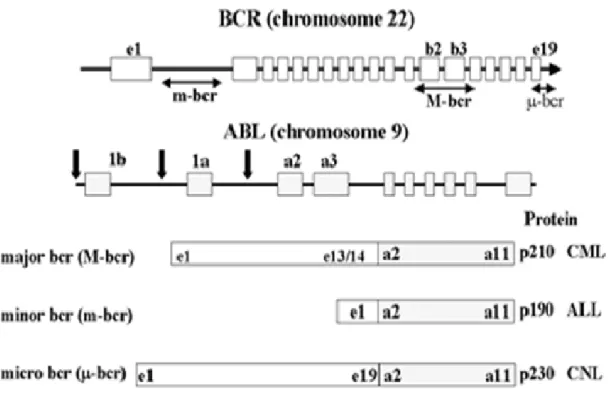
2.2.1.4. BCR-ABL Füzyon Geni ve Füzyon Proteini
BCR ve ABL genlerinin özel bölgelerinden kırılması ve birleşmesi sonucu BCR-ABL füzyon geni oluşur. ABL1 geninde kırık 5’ ucunda 300 kb’lık bir bölgede herhangi bir yerde olusabilir. Genellikle alternatif 1. ekzonlar arasındaki intronda olusur. Ancak BCR-ABL1 mesajcı ribonükleik asit (mRNA) transkriptinde ilk ekzon yoktur ve hep a2 ekzonu ile baslar [18, 19].
BCR geninde kırık 3 farklı noktada olabilir (Şekil 5).
1. KML hastalarının %95’inde ve Ph+ ALL hastalarının yaklasık 1/3-de kırık Majör BCR (M-BCR) denilen bölgede olusur ve sıklıkla ya 13. ekzondan (e13-b2) ya da 14. ekzondan (e14-b3) sonraki introndadır. Sonuç olarak olusan hibrid transkript e13a2 (b2a2) ya da e14a2 (b3a2) birlesimi içerir. Her iki durumda da 210 kd ağırlığında bir füzyon proteini olusur ve p210BCR-ABL olarak adlandırılır [18, 19]. 2. Ph+ ALL hastalarının 2/3- de, KML ve AML hastalarının çok az bir kısmında
görülen diğer BCR kırık noktası minör BCR (m-BCR) denilen bölgede 1. Ekzondan hemen sonra olusur. Sonuç olarak oluşan hibrid transkript e1a2 birlesimini içerir ve 190 kd ağırlığında bir protein kodlar ve p190BCR-ABL adlandırılır [18, 19].
3. KML olgularının çok az bir kısmında ve kronik myeloproliferatif hastalıklar sınıfından kronik nötrofilik lösemi (nötrofilik KML) hastalarında görülen üçüncü
9
kırık noktası mikro BCR (µ-BCR) olarak adlandırılır ve 19. ekzondan (c3) sonra olusur. Hibrid transkript e19a2 birlesiminden olusur ve 230 kd ağırlığındaki p230BCR-ABL proteinini kodlar [18, 19].
BCR-ABL füzyon geninin kodladıgı 210 kD’luk sitoplazmik onkoprotein; artmıs tirozin kinaz aktivitesine sahiptir ve hücre döngüsü kontrolünden bagımsız olarak hareket ederek lösemiye neden olur.
Şekil 5. Bcr ve Abl genlerindeki kırılma noktaları ve Bcr-Abl füzyon proteinleri
2.2.2. KML’DE MOLEKÜLER PATOGENEZ:
2.2.2.1 BCR-ABL1 Aracılı Lösemik Transformasyonun Moleküler Mekanizması:
BCR-ABL füzyon proteini malign transformasyonunu 4 temel mekanizma ile gerçeklestirir. Bu mekanizmalar hücrelerin adhezyon özelliklerini kaybetmesi, mitotik
10
çoğalmayı sağlayan sinyallerin sürekli aktif olması, apoptozun engellenmesi ve proteazomların ABL gen aktivitesini engelleyen proteinleri parçalamasıdır (Şekil 6)[20]
Şekil 6: BCR-ABL geni tarafından kontrol edilen malign transformasyon
mekanizmaları
Apoptozis
Apoptozis genetik olarak programlanmış hücre ölümü olarak tanımlanır ve belirli bir hücre klonunun artmasını sınırlar. İnsan modeli hücre hatlarında BCR-ABL ekspresyonunun tirozin kinaz aktivitesi ve Ras onko geninin aktifleşmesi ile büyüme faktörü eksikliğinden kaynaklanan apoptozu engellediği saptanmıştır [21]. Bundan başka BCR-ABL pozitif hücre hatlarında DNA’da oluşan zararlardan kaynaklanan apoptoza karşı bir direnç geliştiği izlenmiştir. Bcl-2 ve Bcl-xL, ana olarak mitokondri dış membranı, endoplazmik retikulum ve çekirdek zarının sitosolik yüzeyinde yer alırlar ve apoptozu engelleme fonksiyonunu ya kaspazların öncü formlarını durdurarak ya da kaspaz akışını direkt olarak aktive eden sitoplazmadaki apoptoz uyarıcı faktör ve sitokrom-C gibi apoptojenik faktörlerin mitokondriden serbestleşmesini engelleyerek gerçekleştirir [22, 23]. BCR-ABL füzyon proteini, Bcl-2 geninin ekspresyonunu Ras ve/veya PI-3 kinaz yolu ile kontrol eder. Ayrıca BCR-ABL pozitif hücrelerde STAT5’in
11
aktivasyonu ile anti apoptotik bir protein olan Bcl-xL’in transkripsiyonel olarak aktif hale geçtigi belirlenmiştir [20, 24].
BCR-ABL proapoptotik Bad proteinini serin aminoasidinden fosforile ederekte apoptozu engellediği gösterilmiştir. Akt’nin yanı sıra Raf-1 proteinini de serin aminoasidinden fosforile eder [25]. BCR-ABL nin hücrelerin çoğalmasını tetikleyip apoptozu engelleyen mekanizmalar tam anlamıyla anlasılamamıstır. BCR-ABL, hücre içi dengeyi hücrenin büyümesi yönüne çevirirken apoptozuda engeller [20].
Hücresel Adezyonun bozulması
Normal hematopoezde öncül hücreler kemik iliği stroma hücrelerine ve ilgili ekstraselüler matrikse tutunur. Adezyon molekülleri, hücrelerin özgül olarak dokulara yönlenmelerinde, birbirlerini tanımalarında, embriyogenez, hücre büyümesi, hücre farklılaşması ve inflamasyonun düzenlenmesinde görev alırlar. KML öncül hücrelerinin kemik iliği stroma hücrelerine ve ekstraselüler matrikse adezyonu azalır [26]. BCR-ABL, kemik iliğinden hematopoetik kök hücrelerin normal olmayan serbestleşmesiyle sonuçlanan tutunma ve bağlanma bağımsızlığını tetikler [27]. KML öncül hücrelerinin kemik iliği stroma hücrelerine ve hücre dışı matrikse adezyonu azalır. Stromaya adezyon hücre proliferasyonunu negatif olarak düzenler ve KML hücreleri değisen adezyon özellikleri ile bu düzenden kaçıs gösterirler. Son çalısmalar stroma ile öncül hücreler arasında ki etkileşimde β- integrinlerin önemli rolü olduğunu göstermistir. KML hücreleri normal öncül hücrelerde bulunmayan β 1-integrinin adezyon inhibitör varyantını ekprese ederler.
2.2.2.2.
Lösemik dönüşümde rol oynayan sinyal ileti mekanizmaları:
KML ve ALL etyopatolojisinde yer alan BCR-ABL füzyon geni ve buna bağlı mitojenik sinyal yollarının aktivasyonu bugün için aydınlatılmış durumdadır (Şekil 7). BCR-ABL füzyon geni neticesinde tirozin kinaz aktivitesi ile Ras, Raf, PI3K, JNK/SAPK, Crkl ve STAT5 aktivasyonu ile, apopitozis inhibe olmakta ve proliferasyon
12
uyarılmaktadır. BCR-ABL füzyon proteini, üzerindeki tirozin rezidüleri sayesinde daimi bir fosforilasyona ve aktivasyona gitmekte ve Cbl, Crkl, Shc, Grb2 gibi adaptör proteinlerin devreye girmesine ve Ras, PI3K, paxillin ve talin gibi fokal adezyon moleküllerinin, diğer sinyal ileti sistemlerinin aktivasyonuna yol açmaktadır [28].
Şekil 7: BCR-ABL füzyon geninin etkili oldugu yolaklar
Ras/Raf/MEK/ERK ve MAP Kinaz Sinyal İletim Yolu:
Hematopoetik hücrelerde proliferasyonu indükleyen ve apopitozu engelleyen büyüme faktörleri ve sitokinlerin aktive ettiği Ras yolağı, lösemide önemi gösterilmis bir sinyal iletim sistemidir (38). BCR-ABL, Ras sinyal yolağını aktive eden proteinlere direkt bağlanabilir. Ras’ın aktivasyonu Ph kromozomu pozitif lösemilerin patogenezinde önemlidir. IL-3 gibi sitokin reseptörlerinin uyarılması Ras’ın aktivasyonuna neden olur ve aktif Ras proteini serin-threonin kinaz Raf’a bağlanır ve Raf proteini hücre membranında toplanır. Burada serin-threonin aminoasitlerinden fosforillenen Raf proteini aktif hale geçer. Raf, gen transkripsiyonunun aktivasyonuna
13
neden olan Mek1/Mek2 ve Erk üzerinden sinyal yolağını baslatır. Bundan baska Jnk/Sapk yolağının BCR-ABL tarafından aktivasyonu üzerinde çalısılmıs ve malign transformasyon için gerekli olduğu görülmüstür. Böylece Ras üzerinden gelen sinyal GTP-GDP değisim faktörü Rac tarafından Gckr (germinal center kinase related)’ e iletilir ve buradan da Jnk/Sapk yolağı aktive edilir. MAP kinazlar, “Mitogen-activated protein kinases” süper ailesinde yer alırlar. Ökaryotik hücrelerin tümünde mevcut olan bu proteinler hücre membranından çekirdeğe bilgi aktarılmasında önem taşımaktadır. Bu sinyal iletimi kaskadları, embriyogenezis, yaşama, çoğalma, diferansiasyon ve apopitozis işlevlerinin düzenlenmesinde rol alır. [29-33]
PI-3 kinaz/Protein Kinaz B Sinyal İletim Yolu:
Fosfotidilinositol-3 (PI-3) kinaz enzim aktivitesi Ph pozitif hücrelerin çogalması için gereklidir.. BCR/ABL füzyon proteini; Cbl, Crk ve Crk1 gibi adaptör moleküllerle kompleks oluşturarak PI-3 kinazı aktif kılar [29]. PI3 kinaz yolağının aktivasyonu bir serin-threonin kinaz olan Akt kaskadını aktifleştirir. Akt(Protein Kinaz B olarak da bilinir), IL-3 reseptörünün hedef molekülü olup, pro-apoptotik Bad proteinini hedeflemektedir. Fosfat baglanan Bad proteini, sitoplazmik 14-3-3 proteinleri ile birleserek BCL-XL gibi anti-apoptotik proteinlere baglanamadıgı için inaktif hale gelir [26].
Myc Sinyal İletim Yolu :
Etkili bir transkripsiyon faktörü olarak bilinen Myc geninin farklı malign kanserlerde aşırı ifade edildiği rapor edildiği bilinir. Myc geninin aktivasyonu, BCR-ABL geninin SH2 alanı tarafından sağlanıp kontrol edilmektedir. SH’ domaninin Myc geninin aktivitesini hangi yolaklar aracılığıyla kontrol ettiği bilinmemektedir [26].
JAK-STAT Sinyal İletim Yolu:
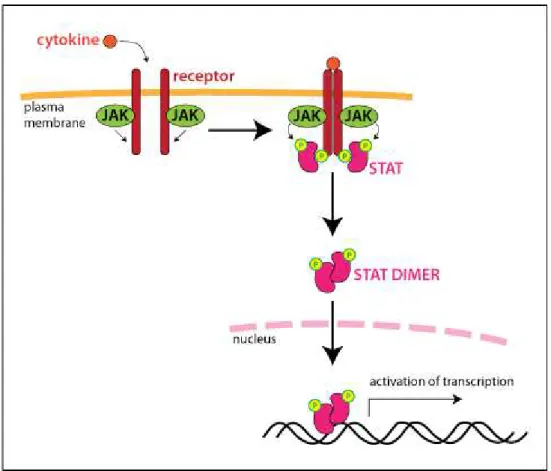
Sinyal ileticisi ve transkripsiyon aktivatörü (signal transducer and activator of transcription; STAT) proteinleri, sitokin, büyüme faktörü veya peptid reseptörlerinden aldıkları sinyaller ile aktive olan ve bu sinyalleri çekirdeğe ileten sessiz (latent) sitoplazmik transkripsiyon faktörleri olarak işlev görürler [34]. Sitokin dönüştürücü sinyallerin en iyi bilinen şekli bir enzim olan Janus Kinaz (JAK)enzimleri inaktif halde Tip 1 ve Tip 2 sitokinlerinin sitoplazmik uçlarına tutunmuştur. Sitokin molekülünün
14
bağlanması ile iki reseptör molekülü bir araya gelir, reseptör ilişkili JAK fosforilasyon yoluyla aktive olur, sonrasında reseptörlerin sitoplazmik bölgelerindeki tirozin rezidülerini fosforile ederler. Reseptörlerin bazı fosfotirozin alanları reseptörüne tutunan monomerik, sitozolik STAT proteinlerinin Src homolog 2 (SH2) uçları tarafından tanınır. Daha sonra STAT proteinleri reseptörle uyarılan JAK kinazlar tarafından fosforile edilirler. STAT proteinlerinin SH2 ucu başka bir STAT proteininin fosfotirozin rezidüsüne bağlanma yeteneğine sahiptir. Sonuç olarak iki STAT proteini birbirine bağlanır ve reseptörden ayrılırlar. STAT dimerleri nükleusa göç eder, sitokine cevap veren genin promotor alanlarındaki DNA sekanslarına bağlanır ve gen transkripsiyonunu aktive ederler. Her bir döngüden sonra yeni STAT proteinleri sitokin reseptörlerine bağlanabilir, fosforilize olabilir, dimerize olup tekrar nükleusa göçebilir (Şekil 8).
Şekil 8: JAK/STAT Sinyal İletim Yolağı:
Memelilerde 4 tip JAK bulunmaktadır. Bunlar JAK1, JAK2, JAK3 ve Tirozin Kinaz 2 (Tyk2)’dir. Sitokinlerin ilgili reseptöre bağlanması ile ilgili JAK aktive olur.
15
Daha sonra aktivasyon uygun STAT molekülünün uygun promotor bölgeye bağlanması ile devam eder. STAT molekülleri 7 adettir. Bunlar STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B ve STAT6 olrarak isimlendirilir (Şekil 9). Farklı birçok grup sitokinlerce ve hematopoetin ile aktive olurlar.
Şekil 9: STAT moleküllerinin yapıları
STAT3 ve STAT5 aktivitelerinin kontrolsüz işleyişi malign transformasyonda rol oynamaktadır.STAT3 ve STAT5, kontrolsüz çoğalan, apoptoza uğramayan, immun sistemden kaçan ve anjiogenezi uyaran tümör hücrelerinde yüksek oranda ifadelenirler [34]. STAT proteinleri iki mekanizma aracılığıyla karsinogenezde etkili olur. Bunlardan biri STAT’ın sürekli aktivasyonudur. Diğer değişim ise proteinin c-ucunun mutasyona uğramasıdır. Devamlı olarak aktif olan STAT proteini antiapopitotik yolları uyararak malign süreçte etkili olabilir. STAT proteinlerinden özellikle STAT3 ve STAT5 kontrolsüz çoğalan, apoptoza uğramayan, immun sistemden kaçan ve anjiogenezi
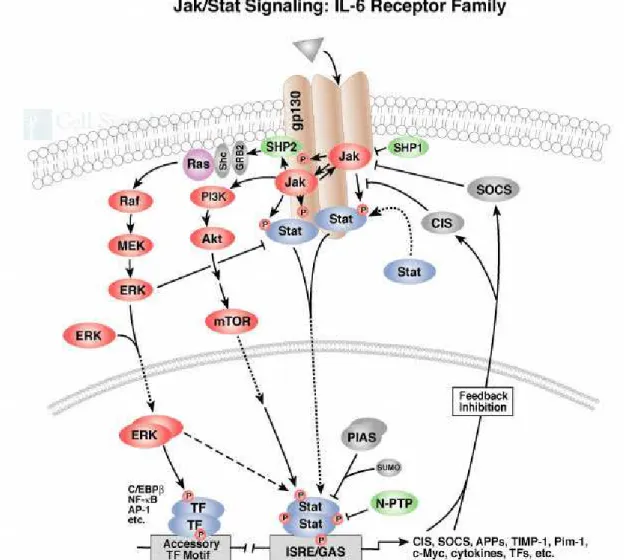
16
uyaran tümör hücrelerinde yüksek oranda ifade edilirler. IL-6 ile JAK-STAT yolağı birkaç yolakdan aktive olmaktadır (Şekil 10). IL-6 ile devamlı STAT3 aktivasyonu uyarılan multipl myeloma hücrelerinde antiapopitotik proteinlerden Bcl-xL ve Mcl-1’in arttığı gösterilmiştir [26]. STAT aracılı sinyal iletimi ile uyarılan hedef genlerin (örneğin; c-myc, siklin D1 ve Bcl-xL) hücre döngüsünün kontrolünü sağlayarak ve/veya apoptozisi önleyerek karsinogenez sürecinde etkili oldukları öne sürülmektedir. Ayrıca, STAT aracılı sinyal iletiminin malign sürecin gelişiminde MAP kinaz yolunun aktivasyonu ile etkileşebileceği düşünülmektedir.
17
Lösemik blastlarda sitokin sentezi ve otokrin/parakrin yolla JAK-STAT yolunun uyarılması, akut miyeloid lösemide devamlı STAT aktivasyonu nedeni olabilecek mekanizmalar arasında sayılmaktadır. STAT3 aktivitesi vasküler endotelyal büyüme faktörü (VEGF) düzeyinin artışına yol açarak, tümör anjiyogenezisinde de rol oynamaktadır. BCR-ABL kimerik proteini hematopoetik hücrelerde büyüme faktöründen bağımsız olarak çoğalma ve transformasyonu indükler. Bu onkoprotein JAK-STAT yolunun sürekli aktif olmasına yol açar.
JAK-STAT yolağının çok sayıda negatif regülasyon mekanizmaları belirlenmiştir. Sitokin Sinyali Baskılayıcısı (SOCS) olarak bilinen proteinler STAT yolağı inhibitörleridir. SOCS STAT'ların negatif düzenleyicisi olarak JAK kinaz reseptörlerine bağlanıp STAT aktivasyonunu baskılar [35, 36]. Diğer bir negatif düzenleyici ise aktive STAT’ ların protein inhibitörleri (protein inhibitors of activated STATs; PIAS)’dir ki bunlar doğrudan STAT proteinleriyle etkileşirler [37]. STAT’lar çeşitli nükleer proteinler ile etkileşerek de transkripsyonu düzenleyebilirler. Bahsedilen tüm bu moleküller potansiyel ilaç hedeflerini oluşturmaktadırlar. İmatinib mesilat ile tetiklenen apoptozis STAT5 aktivitesinin inhibisyonu ve Bcl-xL ekspresyonunun azalması ile korelasyon göstermiştir. Buna göre BCR-ABL ile ilişkili apoptozis direncinde STAT5 aktivitesinin rolü vardır [38, 39].
Çalışmamızda temel olarak STAT5 alındığı için STAT5A ve STAT5B üzerinde özel olarak durulacaktır.
2.2.2.3. STAT5’in genel özellikleri:
STAT5A, meme bezi gelişimi için gerekli, prolaktin sinyal iletiminden de sorumludur; STAT5B ise, büyüme hormonu sinyal iletiminde etkilidir [40, 41]. Her iki STAT5 geninin delesyonu, hematopoietik gelişim bozukluğuna neden olmaktadır [40, 42]. İnterlökin 3 (IL3), granülosit makrofaj koloni stimüle edici faktör (GM-CSF), granülosit koloni stimüle edici faktör (G-CSF), eritropoietin (EPO), trombopoietin (TPO) gibi hematopoietik hücre proliferasyonunu ve gelişimini uyaran çeşitli hematolojik sitokinler, hormonlar ve büyüme faktörleri STAT5’leri aktive edilebilir
18
[40]. STAT5 proteinlerinin, tümör hücrelerindeki ifadelenmelerinin baskılanması apoptozu başlatabildiklerinden dolayı bu proteinlerin kanser tedavisinde önemli birer terapötik hedef olabileceği düşünülmektedir.
STAT3 ve STAT5’in birçok değişik tümör tipinde BCL-XL ifadelenmesi ile hücre apoptozunu düzenledikleri gösterilmiştir [39, 43, 44]. BCL–2 ailesinin bir diğer anti-apoptotik etkili üyesi MCL1 de hem STAT3 hem de STAT5’in hedefidir. Tümör hücrelerinde bu STAT’lardan herhangi biri baskılanırsa, MCL1 ifadelenmesinin azaldığı ve tümör hücresinde apoptozunun başladığı bildirilmiştir [45, 46].
STAT3 ve STAT5’in her ikisinin de kontrolsüz çoğalma ve sağ kalımla ilgili genleri dolaylı veya doğrudan etkiledikleri gösterilmiştir. STAT3’ün anjiogenez ve immun sistemden korunma üzerindeki bilinen etkileri STAT’ları ideal bir kanser tedavi hedefi yapmaktadır [32].
2.3. KML’de Tanı:
Tanıda ilk aşamada ayrıntılı öykü , fizik muayene, tam kan sayımı, biyokimya tetkikleri yer alır. Tanı anında hastaların çoğu kronik evrededir. İlk başvuruda hastalar asemptomatik ola bildiği gibi nadiren kanama, trombozlar ve lökostaz sonrası gelişen multiorgan yetmezliği tablosu ile başvurabilirler. Lökositoz KML’nin en tipik bulgusudur. Lökosit sayısı 15.000–500.000/µl arasında değişir. Ancak hemen daima 25.000/µl üzerindedir ve olguların yarısında 100.000/µl’nin üzerindedir. Tanı anında olguların 1/3’ü ile 1/2’sinde trombositoz saptanmasına rağmen, trombosit değeri normal, yüksek veya azalmış olabilir. Ancak trombositopeni mevcutsa öncelikle akselere ve blastik faza geçiş düşünülmelidir. Kronik faz KML'de blast oranı % 3'ün altında iken akselere faz ve blastik fazda bu oran sırasıyla %1-20 ve > % 20 arasında değişir. Bazofil ve eozinofil sayısında artış kötü prognoz belirleyicisidir.(Şekil 11). Tanı kanda, kemik iliği aspirasyon ve biyopside sitogenetik ve polimerize zincir reaksiyonu (PCR) ile Ph kromozomu ve BCR-ABL’nin gösterilmesi ile konur.
19
Şekil 11. KML, periferik yayma
A) Kronik evrede trombosit sayısı yüksek olabilir, artmış trombosit ve granüler seri elemanlar
B) Hızlanmış evre. Bazofil parçalılarda aşırı artış hızlanmış evre bulguları arasında yer alır. Miyeloblast (myb). Başlangıçta olmayan bir trombositozun gelişmesi de hızlanmış evreye işaret eder (ok).
C) Akut (blastik) evre. Miyeloblastik dönüşümün başladığı görülüyor. Olgun myeloid elemanların yanında blastik hücreler var (oklar).
Standart bantlama yöntemi ile yapılan sitogenetik analizde KML olgularının %90-%95'de Ph kromozomu pozitif olarak gösterilmektedir [t(9;22)(q34; q11)]. Ph kromozomu gösterilmeyen KML düşünülen olgularda ek moleküler yöntemler ile (floresan insitu hibridizasyon [FISH], southern blot ve ters transkriptaz polimerize zincir reaksiyonu [RT-PCR]) BCR-ABL yeniden yapılanması saptanabilir. Bu bulgunun normal sitogenetik analizle bulunmaması genellikle küçük bir bölgenin translokasyonu yönünde değerlendirilebilir. Olgularda takip sırasında yeni kromozomal anomaliler ortaya çıkabilir, bu progresyon ile ilşkilidir [47-50].
2.4. KML’de Ayırıcı Tanı
Lökomoid reaksiyon, myeloproliferatif ve myelodisplastik sendromlar KML ayırıcı tanısında yer almaktadır. Blastik veya akselere fazdaki hastalar akut lösemiler ile karıştırılabilir [51].
20 2.5. KML Klinik evreleme ve prognoz
KML klinik olarak üç evreye ayrılır. Kronik faz, akselere faz ve akut faz (blastik kriz fazı). KML hastalarının %80-85’i kronik fazda, %10’u akselere fazda, %10’u da blastik fazda teşhis edilmektedir. Doğal seyrinde KML tipik olarak kronik faz ile başlar, birkaç yıl içinde akselere faza progresyon olur ve son evre olarak da hastalık blastik faza(blastik kriz) ilerler. Kronik fazdan akselere ve blastik faza geçişin en önemli nedenlerinden biri yeni ortaya çıkan kromozom anomalileridir [52]. Blastik fazda trizomi 8, iso(17q), +19 ve Y kromozomu kaybı, daha az sıklıkta trizomi 19, trizomi 21, kromozom 7 ve 17 gibi ek kromozom anomalileri görülebilir [53, 54]
Kronik faz: Hastaların yaklaşık olarak % 85’i tanı anında kronik fazdadır. Hastalar genellikle asemptomatiktirler veya yorgunluk, karında şişkinlik gibi konstitusyonel semptomlar bulunur. Küratif tedavi yokluğunda hastalık kaçınılmaz olarak diğer fazlara ilerler. Periferik kandaki beyaz kürelerde veya kemik iliğindeki hücrelerde myeloblast oranı tipik olarak %10’ nun altındadır [52, 55].
Ortalama 3-4 yıllık bir kronik fazdan sonra akselere fazdan geçerek blastik transformasyon görülür. Akselere fazda ateş, kemik ağrıları, kilo kaybı gibi belirtilerle lökositoz, spenomegali, trombositoz veya trombositopeni, bazofili ve lökosit alkalen fosfataz (LAP) skorunda ve blast oranında artma görülür. Bu fazda konstitüsyonel semptomlar çok belirgindir ve dalak daha da büyür, sternal duyarlılık ve kemik ağrıları görülebilir. Derin bir anemi, trombositopeni ve tedaviye direnç ile karakterizedir.
Akselere ve blastik faz DSÖ kriterleri aşağıda verilmiştir. (tanı için kriterlerden en az birisi olmalıdır)
21 Tablo 1. DSÖ KML akselere faz tanımı
• Çevresel kan lökositlerinin ve/veya çekirdekli kemik iliği hücrelerinin % 10-19’unun blast olması
• Çevresel kandaki bazofillerin ≥%20
• Tedavi ile ilişkisiz kalıcı trombositopeni < 100.000/mm3 veya tedaviye yanıtsız kalıcı trombositoz > 1x106 /mm3
• Tedaviye yanıtsız ve giderek artan dalak büyüklüğü ve lökosit sayısı • Sitogenetik olarak klonal dönüsüm olması
Tablo 2. DSÖ KML blastik faz tanımı
• Periferik (çevre) kan lökositlerinin veya kemik iliğindeki çekirdekli hücrelerin ≥% 20’sinin blast olması
• Kemik iliği dışı (Ekstramedüller) blastik proliferasyon • Kemik iliği biyopsisinde gruplar halinde blastların olması
Blastik transformasyonda prognoz çok kötüdür. Blastik faz birkaç ay sürüp ölümle sonuçlanır.
Günümüzde, tirozin kinaz inhibitörlerinin tedaviye girmesi ile birlikte blastik faz çok daha nadir görülmektedir. Tedaviye direnç gelişimi de prognozla ilişkili
bulunmuştur [56].
Tedavide imatinib kullanımından önce, KML kronik faz hastalarında prognostik risk değerlendirmesi temel olarak Sokal skoru ve Hasford (Euro) skoru olarak
isimlendirilen iki sisteme dayandırılmıştır ve tedavi öncesi değerler dikkate alınmaktadır.
22 Tablo 3. KML Risk Skorlamaları
I-Sokal risk skoru Hesaplanması Risk Sınıflandırması
Düşük risk < 0.8 Orta risk 0.8-1.2 0.0116 x [yaş (yıl) - 43.4)] + 0.0345 x (dalak -7.51)
+ 0.188 x [(trombosit sayısı/700)2 - 0.563] +0.0887 x (blast - 2.10)
Yüksek risk >1.2
II-Euro, Hasford risk skorlaması (modifiye Sokal) Risk Sınıflandırması
Düşük risk ≤780 Orta risk 781-1480 Yaş≥50 ise 0.666 +(0.042 x dalak boyutu) + trombosit sayısı > 1500 x109/L ise 1.0956 + (0.0584 x blast +bazofil
sayısı > %3 ise 0.20399 + (0.0413 x eozinofiller)x100 Yüksek risk >1480
Not: Dalak büyüklüğü kot kavsinden en uzak nokta (cm) olarak alınır, blast, bazofil ve eozinofiller periferik kandaki yüzdelerdir. Tüm bu faktörler herhangi bir tedavi başlanmasından önceki değerlerdir [57].
2.6. KML Tedavisi:
Amaç öncelikle hastalığa ait semptom ve bulguları kontrol altına almak ve sonrasında kür sağlamaktır. Lökosit sayısı çok yüksek ve lökostaz/hipervizkozite sendromu bulguları varsa lökoferez uygulanabilir [56].
KML tedavisinde başlangıçta hastalığın biyolojik seyrini değiştirmeyen hücre sayısını azaltıcı sitotoksik tedaviler (başlıca hidroksiüre ve busulfan) kullanılmıştır. Günümüzde ise hidroksiüre, lökostatik komplikasyonları önlemek amacıyla tirozin kinaz inhibitörlerinin kullanılmasına kadar kısa süreli kullanılabilir. Ph kromozomu üzerine etkisi yoktur [56, 58]. 1980’li yıllarda ise İnterferon (IFN) ve IFN/ ARA-C kombinasyonu kullanılmaya başlanmıştır. Hastalığın seyri ve kaderi 1998 yılında tirozin kinaz inhibitörlerinin tedavide kullanılmaya başlanılmasıyla değişmiştir. İlk tirozin kinaz inhibitörü olan imatinib mesilat KML tedavisinde bir dönüm noktası olmuş ve ardından direnç gelişimi üzerine ikinci kuşak tirozin kinazlar geliştirilmiştir. Ancak günümüz bilgileri ile tüm gelişmelere rağmen bilinen tek küratif tedavi allojeneik kök hücre naklidir; ancak mortalite ve morbiditesinin yüksek olması nedeniyle tirozin kinazlara dirençli olgular dışında tedavideki yeri oldukça azalmıştır [59].
23
Tablo 4: ELN 2013 Kılavuzuna göre Kronik Myeloid Lösemi Tedavisi [60]
Hat TKİ standart dozu1 transplantasyon
Kronik faz araştır Allo-KİT İm at in ib 4 0 0 M g/ gü n N ilo ti n ib 2 x3 0 0 m g/ gü n D as at in ib 1 0 0 m g/ gü n B o su ti n ib 5 0 0 m g/ gü n P o n at in ib 4 5 m g/ gü n H LA - d o ku u yu m u A kr ab ad ış ı d o n ö r d ü şü n ü le n ö n e ri le n ke m o te ra p i 1. Bazal X X X X2
1. TKİ’e intolerans 1. hat onaylı Herhangi başka TKİ imatinib X8 X X X X nilotinib X X X X X X 2. dasatinib X8 X X X X X
3. 2 TKI karşı intolerans
ve ya yanıtsızlık Kalan herhangı TKİ
H e rh an gi T315I mutasyonu X X X X
Akselere veya blastik faz Başla X3
X4 X X
Yeni tanı ,
TKİ naif hasta Optimal Yanıt yoksa
X7 X5
Daha önce
TKI alan hastalar Herhangi başka TKİ X
6 X7 X5
1)-TKİ seçimi hastanın tolerabilitesi , güvenirliği ve karakteristik özellikleri (yaş , komorbid durumlar ) göz önünde bulundurularak yapılmakta ; 2)-sadece 1 temel uyarı varlığı halinde (yüksek risk , majör KKA/Ph+ ; 3)-2x400 mg/gün, 4)- 2x70 mg/gün veya 140 mg/gün, 5)-hastalığı kontrol altında tuta bilmek için nakil öncesinde ve hastanı AlloKİT’e uygun hale getirmek amaçlı ;6)- T315I mutasyonu olan olgularda; 7)- sadece Allo-KİT’e uygun hastalar; 8)-yanıtsızlık durumunda 2x400 mg/gün [60].
24
THD tarafından 2013 yılı KML kılavuzunda tedavi önerileri 2013 yılı ELN kılavuzuna göre belirlenmiştir [61-64].
Tablo 5. THD 2013 kılavuzuna göre kronik evre KML tedavisinde birinci, ikinci ve
daha sonraki basamaklardaki tedavi önerileri [57].
Birinci basamak
İmatinib (400 mg/gün) veya nilotinib** veya dasatinib**
Başlangıçta uyarı (yüksek risk, KKA/Ph+) olan hastalara ve kardeşlerine HLA doku grubu bak
İkinci basamak, ilk TKI’ye intolerans
İlk kuşakta denenmemiş TKI’dan herhangi biri (nilotinib veya dasatinib)
İkinci basamak, imatinibe yanıtsız
Dasatinib veya nilotinib veya bosutinib¶ veya ponatinib§ Hasta ve kardeşlerine HLA doku grubu bak
İkinci kuşak, nilotinibe yanıtsız**
Dasatinib veya bosutinib¶ veya ponatinib§
Hasta ve kardeşlerine HLA doku grubu bak; akraba dışı kök hücre vericisi ara ve allo-HKHN düşün
İkinci kuşak, dasatinibe yanıtsız**
Nilotinib veya bosutinib¶ veya ponatinib§
Hasta ve kardeşlerine HLA doku grubu bak; akraba dışı kök hücre vericisi ara ve allo-HKHN düşün
Üçüncü kuşak, 2 TKI’ye yanıtısız ve/veya intolerans
Kalan TKI lerden herhangi biri; uygun hastalarda allo-HKHN önerilir
Herhangi bir basamak tedavide T315I mutasyonu saptanması
Ponatinib§
Hasta ve kardeşlerine HLA doku grubu bak; akraba dışı kök hücre vericisi ara ve allo-HKHN düşün
**Randomize karşılaştırmalı çalışmalarda nilotinib (ENESTnd) ve dasatinib (DASISION)’in kronik evrede ilk seçenek tedavide imatinib’e göre daha erken ve daha yüksek oranda sitogenetik ve moleküler yanıtlar oluşturduğu gösterilmiştir [61, 62]. Bu verilerin ardından kronik evrede ilk seçenek tedavi için ruhsatlandırıldıkları ülkelerde nilotinib 2x300 mg/gün, dasatinib ise 100 mg/gün dozunda kullanılmaktadır. İlk seçenek tedavide ülkemizde de ruhsatlanmış olmalarına rağmen henüz geri ödeme almamışlardır. Bosutinib ve §ponatinib henüz ülkemizde ruhsatlanmamışlardır. Not-1 : Kronik evrede (ikinci basamak) dasatinib dozu 100 mg/gün, nilotinib dozu 2 x 400 mg/gün’dür. Blastik evrede nilotinib kullanım endikasyonu yoktur. Hızlanmış ve blastik evrede dasatinib dozu 140 mg/gün’dür [63, 64]. Not-2 : Dasatinib ve nilotinibi kaldıramayan hastalarda hastaya göre allo-HKHN, hidroksiüre, interferon, sitozin arabinozid tercih edilebilir.(Çalışma grubunun ortak görüşü)
25
Tablo 6. THD 2013 Kılavuzuna göre Hızlanmış Evre ve Blastik Evre KML
hastalarında tedavi önerileri [57]
TKI almamış yeni tanı Hızlanmış
evre ve Blastik evre hastalar
İmatinib 600-800 mg/gün veya dasatinib 2x70 mg/gün veya 140 mg/gün Allo-HKHN için verici taraması
Blastik evredeki tüm hastalara ve hızlanmış evrede olup uygun yanıt sağlanamayan tüm hastalara allo- HKHN önerilir.
Hastalığı kontrol altına almak için allo-HKHN öncesi kemoterapi gerekebilir.
Daha önce TKI almış Kronik evreden Hızlanmış evre ve Blastik evreye ilerleyen hastalar
Progresyondan önce kullanılmamış herhangi bir TKI (ponatinib§; T315I mutasyonu varsa) ardından tüm hastalarda allo-HKHN
Hastaları allo-HKHN’ye hazırlamak için sıklıkla kemoterapi gereklidir*.
*Genellikle bu hastalarda tek başına TKI kullanılması allo-HKHN öncesinde remisyon sağlamaya yetmeyeceği için sitotoksik kemoterapi uygulanması gereklidir. Kontrolsüz, dirençli blastik evre varlığında allo-HKHN önerilmemektedir. §Ponatinib henuz ülkemizde ruhsatlanmamıştır.
Tablo 7. İkinci kuşak TKİ seçimi [65-67]
Mutasyon Durumuna Göre
Nilotinibe duyarlılığı az olan mutasyonlar Y253H, E255K/V, F359V/C Dasatinibe duyarlılığı az olan mutasyonlar F317L, F317I/V/C, T315A, V299L Nilotinib ve dasatinibe dirençli mutasyon T315I
Eşlik Eden Hastalıklara Göre
Akciğer hastalıkları Nilotinib düşün
Ağır diabetes mellitus, pankreatit öyküsü Dasatinib düşün
QT uzaması olan hastalarda hem nilotinib, hem de dasatinib kullanımı özel dikkat gerektirir.
Not: Nilotinib ve dasatinib’i doğrudan karşılaştıran bir çalışma yoktur. Veriler bu iki TKİ’nin kullanıldığı farklı çalışmalarda izlenen yan etkiler, mutasyon-TKİ direnci ilişkisini yansıtan yayınlar ile uzman görüşlerinin bulunduğu derlemelerden elde edilmiş öneriler niteliğindedir
2.6.1. Tirozin kinaz inhibitörleri
İmatinib, nilotinib (AMN-107), dasatinib (BMS-345825), bosutinib (SKI-606),
ponatinib (AP-24534) ve klinik kullanıma giren birinci ve ikinci kuşak tirozin kinaz inhibitörleri dışında Bafetinib (INNO-406) da üretilmiştir.
26 İmatinib:
Hedefe yönelik olarak geliştirilmiş bir ajan olan imatinib mesilat Platelet-derived growth factor (PDGF) reseptörünü hedef alan spesifik bir tirozin kinaz inhibitörüdür (Şekil 12;13). İmatinib, p210 onkoproteinin ATP bağlanan bölgesine kompetetif inhibisyonla bağlanarak ATP’nin bağlanmasını bloke eder ve böylece ATP’den fosfor transferi engellenerek BCR-ABL proteininin inaktif formda kalmasını sağlar. Sonuç olarak BCR-ABL sinyal iletiminde görevli proteinlerin tirozin fosforilasyonu inhibe olur, bu sayede sinyal ileti yolaklarını bloke ederek hücre büyümesinin duraklamasına ve hücre ölümüne neden olur. İmatinib’in Ph(+) KML’li hastaların füzyon ürünlerinden başka gastrointestinal stromal tümörlerde (GİST) artmış ekspresyonu olan C-kit (CD 117)’i inhibe ettiği de gösterilmiştir. İmatinib sadece BCR-ABL inhibisyonu yapmaz. Aslında etki gücüne göre sırasıyla DDR-1 ve DDR-2> PDGFR-α ve PDGFR-β > c -KIT > BCR-ABL tirozin kinazlarını etkilemektedir [68]. Imatinib, "Food and Drug Administration" (FDA) tarafından Mayıs 2001’de IFN tedavisine refrakter KML tedavisi ve Şubat 2003’te yeni tanı KML tedavisi için onaylanarak kullanıma girmiştir. İmatinib, daha önceki standart tedavilerle karşılaştırıldığında, uzun dönemde yüksek yanıt oranları ve iyi yan etki profili nedeniyle, kronik faz KML’nin standart tedavisi olarak kabul edilmiştir.
27
Tedavi, genel olarak iyi tolere edilir. Yan etkiler arasında kas iskelet şikayetleri, ödem, plevral efüzyon, assit, kilo alımı bulantı, kusma, ishal, makülopapüler cilt döküntüsü, halsizlik, baş ağrısı, doza bağımlı hepatotoksisite ve myelosupresyon sayılabilir[69, 70].
İmatinib tedavisi sırasında rastlanan en önemli sorunlardan biri de imatinib
direncidir.
İmatinib direnci:
Direnç görüldüğü zamana göre primer ve sekonder olarak adlanırılır. Primer imatinib direnci, 3. ayda tam hematolojik yanıt elde edilmemesi, 6. ayda herhangi bir sitogenetik yanıt elde edilememesi, 12. ayda kısmi sitogenetik yanıt elde edilememesi veya 18. ayda tam sitogenetik yanıt elde edilememesi olarak kabul edilirken, hastalığın herhangi bir devresinde tam hematolojik yanıt kaybı, tam sitogenetik yanıt kaybı veya hastalık ilerlemesi durumu, kazanılmış direnç (yanıt kaybı) olarak ifade edilmektedir [71]. İmatinib direnci BCR-ABL bağımlı ve bağımsız olarak tanımlanır.
BCR-ABL bağımlı direnç mekanizmaları başlıca olarak ATP bağlayan domainde
(P-loop) veya ABL onkoprotein’in aktivasyon lupunda noktasal mutasyonlar, BCR-ABL füzyon geninde genomik amplifikasyon, BCR-BCR-ABL yazımında düzensizlik, çoklu direnç proteinlerinin düzensizliği nedeni ile imatinibin hücre dışına atılması ve imatinib’in hücresel biyo-kullanılabilirliğinin azalmasına dayanmaktadır. BCR-ABL’de kinaz bölgesi mutasyonları, kazanılmış imatinib dirençi olan hastaların %50-60’ını oluşturur. İmatinib dirençli KML vakalarında ABL nokta mutasyonlarının gelişmesi yeni tirozin kinaz inhibitörlerinin gelistirilmesine yol açmıstır. İmatinib direnci olan KML hastalarında BCL nokta mutasyonlarının sıklıgı sırasıyla: T315I %15, Y253H %5, E255K %15, F317L %3 olarak bildirilmektedir. T315I mutasyonu, dasatinib ve nilotinib tedavisine dirençlidir. T315I mutasyonu olan olgular için yeni tedavi ajanları geliştirilmektedir ve bu ajanlarla ilişkili faz I ve faz II çalışmalar devam etmektedir. Bu olgularda allojenik kök hücre transplantasyonu önerilmektedir. Y253H, E255K mutasyonlarında dasatinib’in nilotinib'den daha etkili, F317L/V mutasyonunda nilotinib’in dasatinib'den daha etkili oldugu bildirilmektedir [72].
28
BCR-ABL bağımsız mekanizmalar; hücre içindeki imatinib konsantrasyonu ile
ilgilidir,örneğin; ilaç influx ve effluxu, ve SRC kinaz ailesi gibi BCR-ABL den bağımsız sinyal ileti yolaklarıdır [73, 74].
BCR-ABL’den bağımsız mekanizmalar taşıyıcı proteinlerde (OCT 1, MDR 1) ve farmakolojik bariyerlerde (CYP3A 4/5 indükleyiciler) olan değişiklikler, NF-kappa B, SHP-1, p53, RB, p16 mutasyonları, EVI ve myc aşırı ekspresyonu, kromozomal değişiklikler, ek Ph kromozomu kazanımı, src ilişkili Lyn/Hck kinazların aşırı ekspresyonu gibi bazı sekonder genetik değişikliklere bağlıdır [75, 76].
Nilotinib:
Bir aminoprimidin olan Nilotinib, Abl’ye yüksek oranda selektif olup, BCR-ABL kinaz inhibisyonu etkisi imatinib duyarlı KML hücreleri üzerinde imatinibden 20-50 kat, imatinib dirençli KML hücreleri üzerinde ise 3-7 kat daha potent olan ikinci kuşak tirozin kinaz inhibitörüdür [77]. İmatinibe benzer sekilde BCR-ABL1’in ATP-bağlayıcı bölgesine kompetitif inhibisyonla bağlanarak etki eder, ancak bağlanma affinitesi ve ABL kinaz seçiciliği imatinibden üstündür. Nilotinibin inhibe ettiği kinazları araştıran bir farmakodinamik çalışmada nilotinibin etkinlik gücü sırasıyla; DDR-1 > DDR-2 > BCR/ABL (Abl) > PDGFR-α/PDGFR-β > KIT > CSF-1R olarak ifade edilmiştir [68]. Yapılan çalışmalar sonucu imatinibe dirençli olan ve yeni tanı vakalarda etkin olduğu gösterilmiştir [63, 78, 79].
29
Yeni tanı KML hastalarında (ENESTnd -Evaluating Nilotinib Efficacy and Safety in Clinical Trials of Newly Diagnosed Ph+ CML Patients -çalışması) kronik faz KML’li hastalarda ilk seçenek olarak nilotinib ve imatinib’in etkinliği karşılaştırılmaktadır [62]. Bu çalışmada nilotinibin majör moleküler yanıt (MMY) oranları daha yüksek, akselere ve blastik faza geçme oranları anlamlı oranda düşük saptanmış. Yapılan çalışmalar ve elde edilen sonuçlar sonrasıda nilotinib Ağustos 2010’da 1. Basamak tedavi için FDA onayı almıştır . Nilotinib, yan etki profili tolere edilebilen oral bir ajandır. En yaygın grade 3/4 yan etkiler; trombositopeni (%20-33), nötropeni (%13-31), bilirubin yüksekliği (%7) ve serum lipaz yüksekliğidir (%5-15). Diğer sık yan etkileri döküntü, kaşıntı, baş ağrısı, bulantı ve yorgunluktur. Nilotinib ayrıca kardiyak ventriküler repolarizasyonu uzatarak, QT intervalinde doz bağımlı uzamaya neden olur.
Dasatinib:
Dasatinib (BMS-354825) tiyazolkarboksamid yapısında bir tirozin kinaz inhibitörüdür. Abl kinaz bölgesine aktif ve inaktif konformasyonda bağlanarak Bcr/Abl tirozin kinaz ve Src ailesi kinazlarını inhibe eder. PDGFR, C-kit ve Ephrin-A reseptör kinaz’ı da güçlü şekilde inhibe edebilir. Dasatinib, İmatinib’den yaklaşık olarak 300 kat daha potenttir, ancak T315I mutasyonunda etkin değildir [80-83]. Dasatinib BCR/ABL’nin ATP bağlanma bölgesine bağlanarak tirozin fosforilasyonunu ve hücre içi sinyal ileti yolağını inhibe ederek hücre büyümesinde duraklama ve apoptoza neden olur [84]. Dasatinibin DASSASION çalışması ile birinci basamak kullanımda da imatinibe üstünlüğü gösterilmiştir [61, 85, 86]. Dasatinib imatinibi hücre dışına atan ve hücre içi konsantrasyonunu azaltarak etkisini azaltan efflux mekanizmalarına karşı dirençlidir. Bu mekanizma ile imatinibe direnç geliştirmiş olgularda dasatinib etkin olabilmektedir [84]. Genel olarak iyi tolere edilebilir. Yan etkileri arasında kemik iliği supresyonuna bağlı pansitopeni, ödem, sıvı retansiyonu, plevral efüzyon, miyozit, döküntü, bulantı, kusma, kardiyak ventriküler repolarizasyonu (QT aralığı) uzatma potansiyeli sayılabilir [61].
Ponatinib (AP24534) – T315I mutasyonuna da etki edebilen farklı özellikte 3. Kuşak
30
ilaç dirençlerinin %15-20’sinden sorumludur. Ponatinib, Aralık 2012 de rezistan ve intoleran KML hastalarının tedavisinde FDA onayı almıştır [87, 88].
Bosutinib. Dual Abl/Src inhibitörü olup c-Kit ve PDGFR’ lerine karşı sınırlı etkisi
vardır [74]. BCR/ABL için imatinibden 200 kat daha potenttir ve T315I dışında birçok mutasyona karşı aktivitesi gösterilmiştir [89]. Bosutinib, tedaviye dirençli veya intoleran Ph + KML tanılı erişkin hastaların tedavisi için 2012 yılında ABD’de FDA onayı aldı [90-92].
İkinci Kuşak TKİ dirençli olduğu mutasyonlar :
Tablo 8. İkinci kuşak TKI’lerinde mutasyonlara duyarlılık durumları [57] Mutasyon Durumuna Göre:
Nilotinibe duyarlılığı az olan mutasyonlar : Y253H, E255K/V, F359V/C Dasatinibe duyarlılığı az olan mutasyonlar : F317L, F317I/V/C, T315A, V299L Nilotinib ve dasatinibe dirençli mutasyon : T315I
(KML Tanı ve Tedavi Kılavuzu,THD,2013)
2.6.2. Allojeneik Hematopoetik Kök Hücre Nakli
Küratif tedavi allojenik kök hücre naklidir; ancak mortalite ve morbiditesinin yüksek olması nedeniyle tirozin kinazlara dirençli olgular dışında tedavideki yeri oldukça azalmıştır. Özellikle T315I mutasyonu olan ve tüm tirozin kinazlara dirençli olan vakalarda düşünülmesi uygun olacaktır.
2.6.3. KML’de Tedavi Yanıtının Değerlendirilmesi
2013 Yılı European Leukemia Net (ELN) kriterlerine göre KML tedavisine yanıt; hematolojik yanıt, sitogenetik yanıt ve moleküler yanıt olarak incelenmektedir [60].





![Tablo 4: ELN 2013 Kılavuzuna göre Kronik Myeloid Lösemi Tedavisi [60]](https://thumb-eu.123doks.com/thumbv2/9libnet/3040539.2776/36.892.166.798.170.879/tablo-eln-kilavuzuna-gore-kronik-myeloid-losemi-tedavisi.webp)
