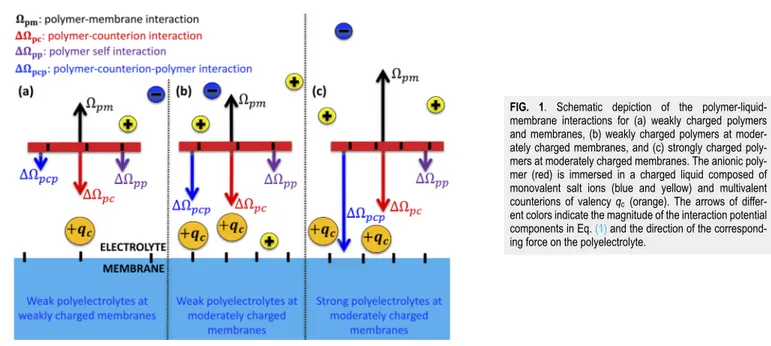
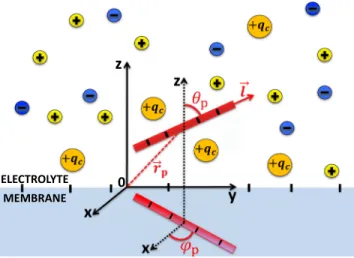
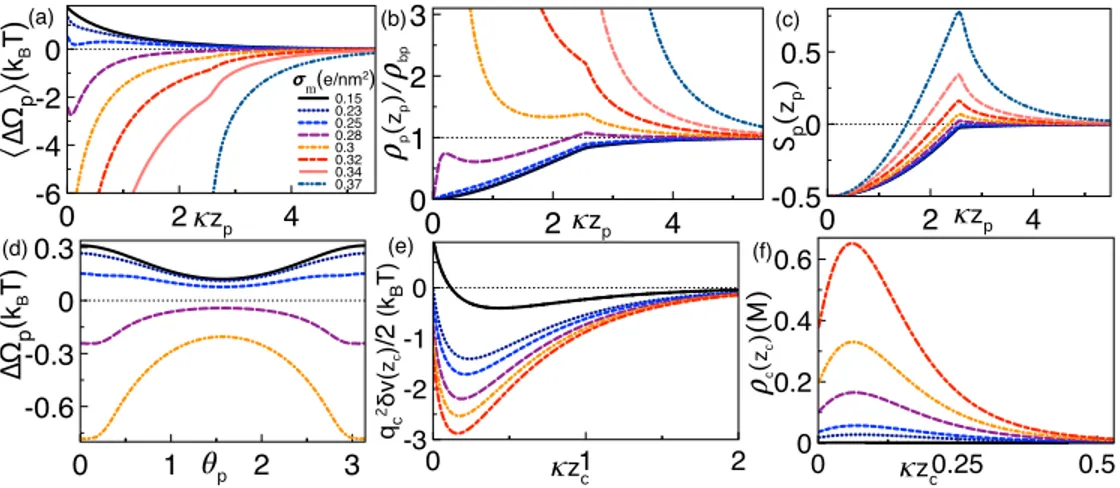
Like-charge polymer-membrane complexation mediated by multivalent cations: one-loop-dressed strong coupling theory
Tam metin
Şekil

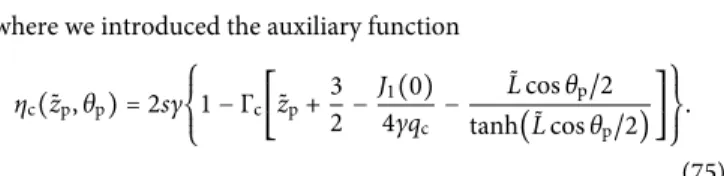
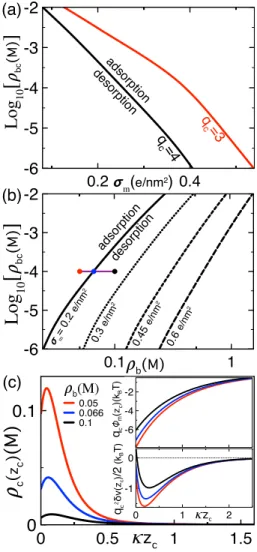
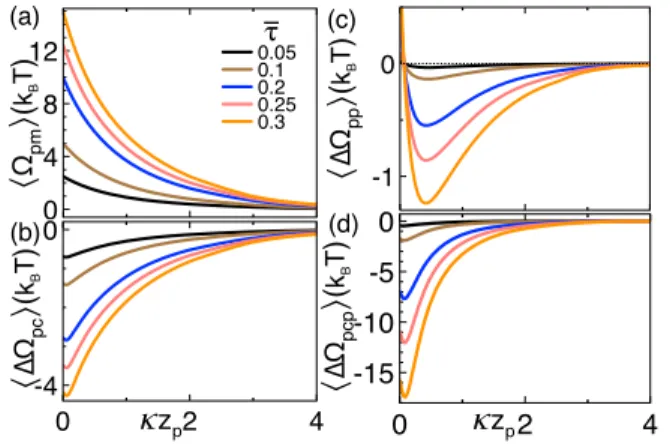
Benzer Belgeler
[email protected] 3 Araş.Gör., Mehmet Akif Ersoy Üniversitesi, Bucak Zeliha Tolunay Uygulamalı Teknoloji ve İşletmecilik
Çabuk ve vd (2015)’de yayınlamış oldukları Büyüyen Yerel Markaların Pazarlama Uygulamalarını açıklayan kitapta, son olarak ele alınan bütün işletmelerin;
Atasözleri ve deyimler sadece geç- miflte yaflanm›fl deneyimleri yans›tan ve sözlü kültüre ait “dil kal›nt›lar›” m›d›r sorusuna yönelik olarak, günlük
Here and hereafter, we use GP, HP, and UP to refer to the respective block-row partitioning methods as well as ABCD Solver that utilizes the regular block Cimmino algorithm for
In this work a multidimensional RF pulse design method is proposed for multi-channel transmit systems which realize the maximum possible SAR reduction with the VERSE operation
Herein, we develop a facile strategy to assemble the vertically aligned monolayer of Au nanorods with a nominal 0.8 nm gap distance and demonstrate their applications in the
什麼情況下要接受根管治療? 返回 醫療衛教 發表醫師 發佈日期 2010/01/29
published a case-controlled study an- alyzing the risk of major coronary events and again found that women with left-sided sided breast cancer had more major coronary events
