Çocukluk cağı lösemi tanılı hastalarda sitogenetik anomaliler
Tam metin
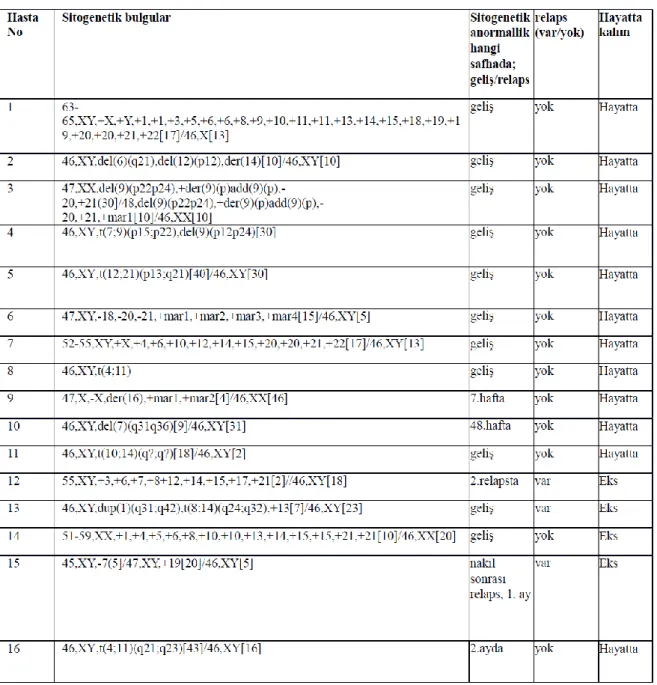
Şekil


Benzer Belgeler
The main factors that affect the unit cost of fabrics which are woven in small enterprises that work on order basis are weaving preparation, raw material, labour, energy and
TNM (tumor-lymph node-metastasis) evrelemesine temel oluşturan larinks bölgelerinin preoperatif olarak doğru değerlendirilmesi larinks kanserlerinin tedavi planlaması,
İlginç olarak RT-PCR ile kemik iliğinden bakılan BCR-ABL t(9;22) pozitif olarak geldi.. Kantitatif BCR-ABL füzyon transkriptinin oranı ise 0.27
İşte bu sebep lerden dolayı binlerce milliyetçi onun peşinden koştu; onun izini kaybetmemek i- çin heyecanları nın dalgalarına kapılarak yorul m adan
Amaç: Bu çalışmada, Ege Üniversitesi Tıp Fakültesi Tıbbi Biyoloji Anabilim Dalı’na 2009-2013 yılları arasında akut myeloid lösemi (AML) ön tanısı ile başvuran 402
Adana’da do¤up büyüyen Devran Baba, bizzat tan›flm›fl oldu¤u Âfl›k Vey- sel’in, bunun yan›nda Âfl›k Ali ‹zzet Öz- kan, Âfl›k Davut Sularî ve Âfl›k
İlk kez 1934 yılında Asbjörn Fölling (1888-1937) isimli Norveçli bir hekim tarafından zihinsel engelli olan sarışın, mavi gözlü iki kardeşte tanımlanmıştır. Hastalık
This theoretical framework represented below explains how neuromarketing sciences when applied on the digital marketing tools like website design, SEO, affiliate
