MeralKoyuturk ,NedretAltiok ,MelikeErsoz SimvastatininducesproliferationinhibitionandapoptosisinC6gliomacellsviac-junN-terminalkinase
Tam metin
Şekil

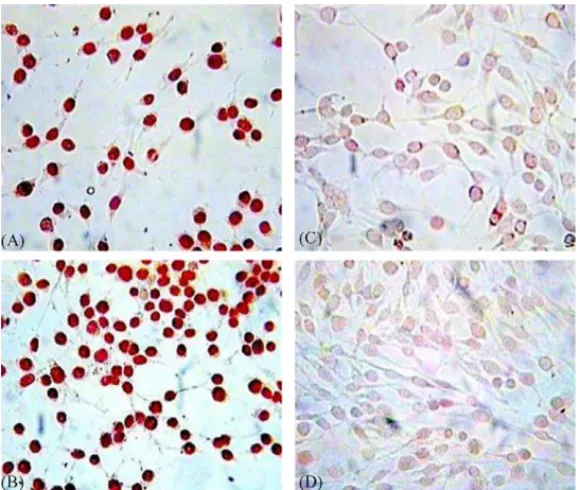
Benzer Belgeler
TPA addition induces the phosphorylation of JNKs and ERKs, but not p38, protein in HL-60 cells, and incubation of HL-60 cells with JNKs inhibitor SP600125, but not ERKs inhibitor,
TPA addition in- duces the phosphorylation of JNKs and ERKs, but not p38, protein in HL-60 cells, and incubation of HL-60 cells with JNKs inhibitor SP600125, but not ERKs
Simülasyonla, Mamdani ve Larsen tip bulanık içermelerin aynı nokta ve aynı açıda karşılaştırılması Şekil 4.8(a) ve 4.8(b)’de, ayrıca, bu iki bulanık
Overexpression of a dominant-negative mutant mitogen- activated protein kinase kinase kinase 1 (MEKK1-DN) or treatment with JNK- specific antisense oligonucleotide or Vit C
We also found that c-Jun NH2-terminal kinase (JNK) but not p38 mitogen-activated protein kinase or extracellular signal-regulated kinase 1/2 was persistently activated in apoptosis
A transient increase in the activation of c-Jun-NH(2)-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) in ECs was observed; however, only ECs pretreated with
We provide evidence that the Jun N-terminal kinase (JNK) signaling pathway mediates Aβ- and ceramide-induced apoptosis: Both Aβ and ceramide activated JNK phosphorylation,
To further examine the role of phosphorylation in HDAg function, two conservative CKII recognition sites at Ser-2 and Ser-123 of both HDAgs and one potential PKC recognition site
