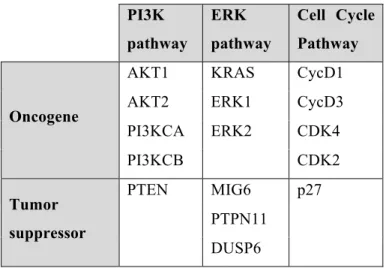
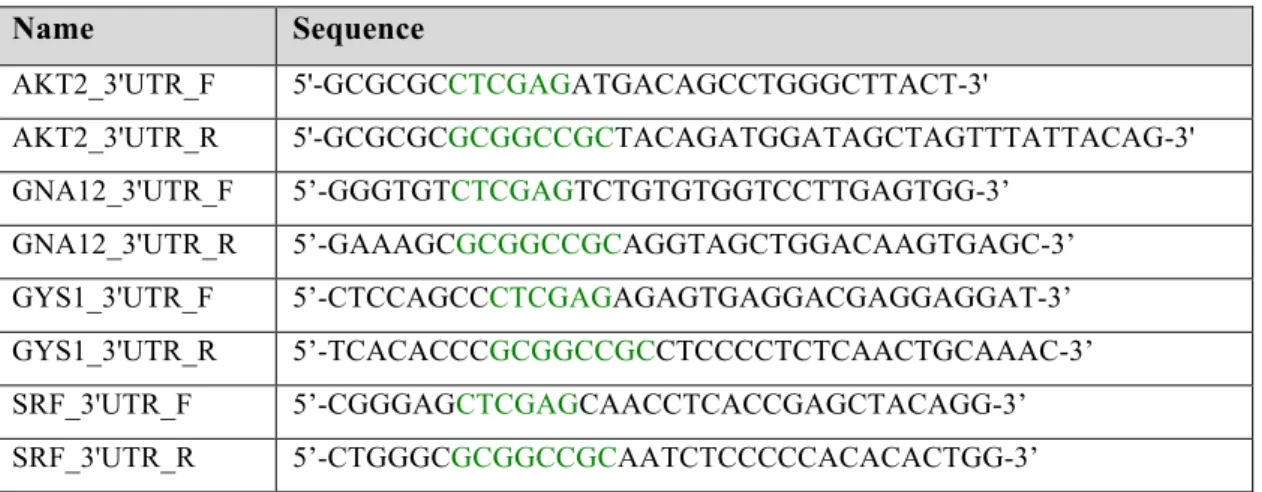
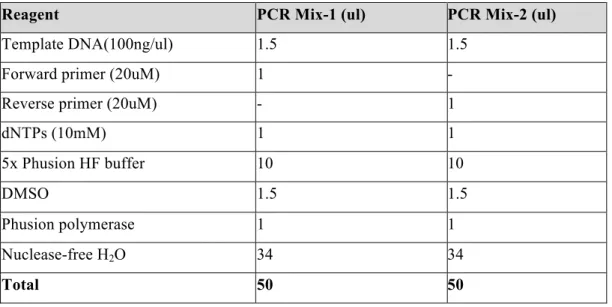
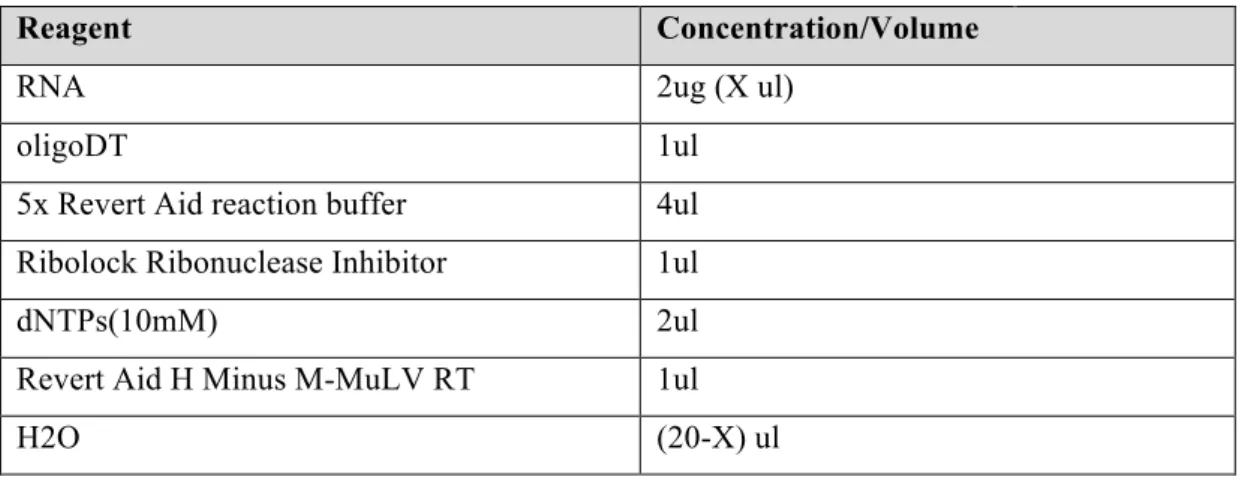
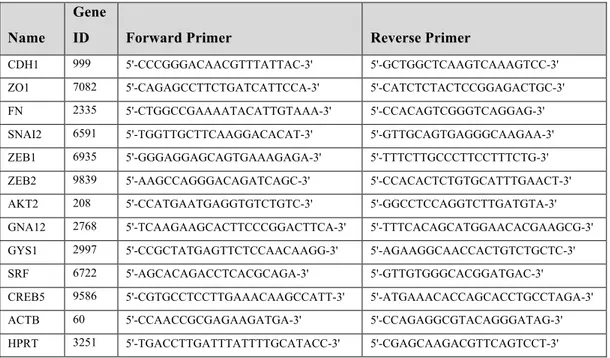
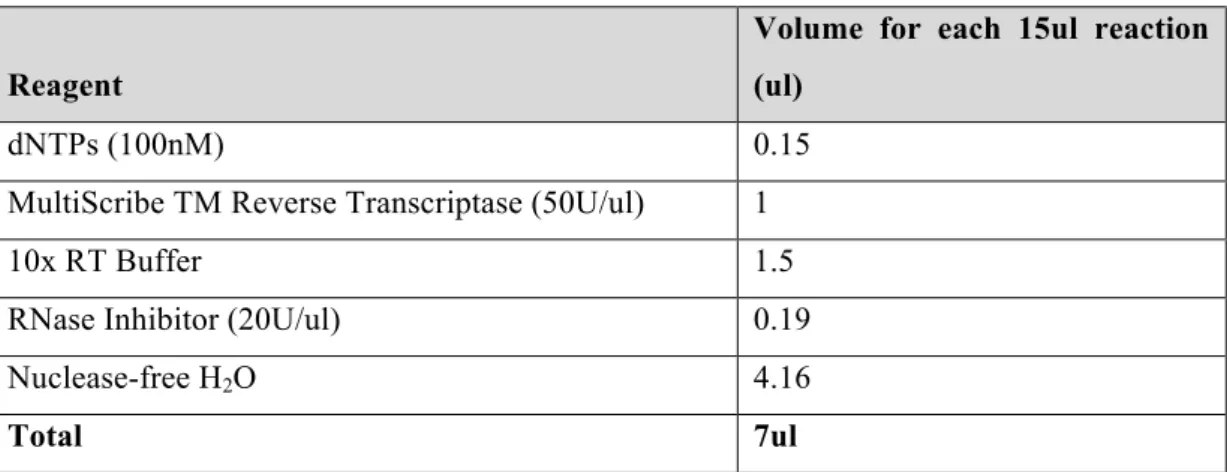
Combinatorial targeting of PI3K and MAPK pathways by miR-564 to inhibit proliferation and invasion in breast cancer
Tam metin
Şekil

![Figure 1.3. Schematic demonstration of miRNA formation and function (Reprinted by permission from Nature Publishing Group: [Nature Reviews Cancer] [33], Copyright (2006).)](https://thumb-eu.123doks.com/thumbv2/9libnet/5636721.111999/20.892.189.649.591.1006/schematic-demonstration-formation-function-reprinted-permission-publishing-copyright.webp)


Benzer Belgeler
Türkiye’de Tarım sektöründe faaliyet gösteren tarımsal kooperatifler 1163 Sayılı Kooperatifler Kanunu ve bu Kanuna değiĢik 3476 sayılı kanuna göre faaliyet gösteren
Jurelang Zedkaia 查凱爾及夫人一行,六月十八日(周 六),蒞臨萬芳醫學中心參訪,並接受健康檢查,查凱爾總統對萬芳先進醫療設備及優質醫療服務留 下深刻印象。
We found that coupling along the external E direction results to shift of the magnetic resonance frequency of the single SRR; the shift is upwards when the neighboring sides of the
Through identity confusion in cultural translation, in the novel “Joy Luck Club” the conflicts between Chinese mothers and their American daughters and the hope of a mother to pass on
Immunohistochemistry for S-100 revealed a moderate to strong positive reaction in the cytoplasm of the brown fat tissue cells (Fig. 3-a), but weak positive immune reaction
The association of clinicopathologic features and peripheral blood parameters with high PD-L1 expression in non-small cell lung cancer Introduction: Programmed death ligand 1
Küçük hücreli dışı akciğer kanseri tanısı olmayan olgular, preoperatif dönemde kemoterapi ve/veya radyoterapi alan olgular, mediastinoskopide mediastinal lenf nodları (N2 ve
Kamu politikası analizi disiplini 1950’lerde Amerika Birleşik Devlet- leri’nde kamu yönetimi ve siyaset biliminden ayrılarak bir inceleme alanı ola- rak ortaya
