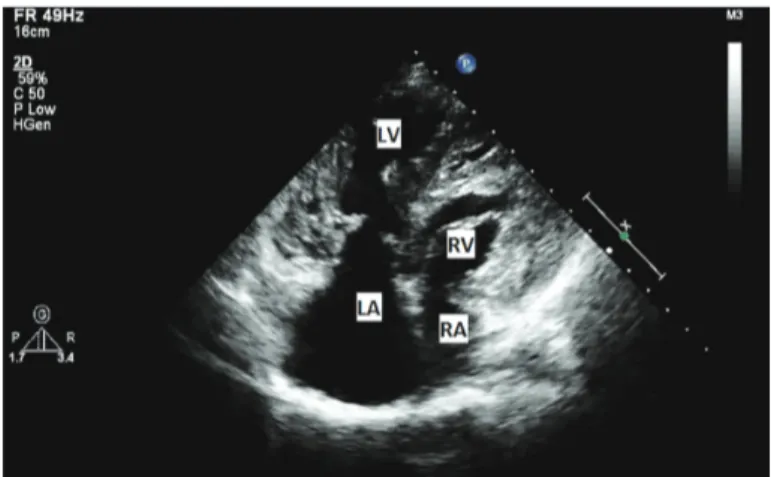
Biventricular non-compaction cardiomyopathy with pulmonary stenosis, interatrial septal aneurysm, atrial septal defect, bradycardia, and mental retardation in a single case: A case report
Tam metin
Şekil
Benzer Belgeler
On the follow-up chest X-ray, we observed an intense, round, homogeneous density, 5x5cm in size, which was absent before the CABG surgery (Fig. Chest computerized tomography revealed
However, the RV involvement with a significant outflow obstruction is uncommon except relatively mild gradient (5–25 mmHg) in RVOT that may occur in some patients with
Operations such as atrial septal defect (ASD) closure with conventional cannulation techniques can be performed safely with minimally invasive RLT method without any
Herein, we present a patient with scimitar syndrome associated with ventricular septal defect (VSD), left pulmonary venous stenosis (PVS), right pulmonary
Surgical treatment of a left atrial rupture during transcatheter atrial septal defect closure: a case study.. Transkateter atriyal septal defekt kapatılması esnasında gelişen
with bronchiectasis that showed concurrent DORV, VSD, pulmonary stenosis (PS), and atrial septal defect (ASD) along with a history of frequent pulmonary infections
We recommend suspecting MTHFR polymorphism in young fertile women with a history of spontaneous abortus or deep venous thrombosis or congenital heart disease with a
Left ventricular outflow tract obstruction can be relieved with a transannular patch in patients with atrial situs inversus with congenitally corrected transposition of
