INSTITUTE OF NATURAL AND APPLIED SCIENCES
Asymmetric Transfer Hydrogenation of Ketones with
Ru (II) and Ir (III) Phosphinite complexes
Ahmed SABRI
M.Sc. Thesis
DEPARTMENT OF CHEMISTRY
TURKEY - DİYARBAKIR JANUARY 2016
I
“ACKNOWLEDGEMENTS”
“First of all, I would like to express my”cordial thanks“to my supervisor,”Assoc. Prof. Murat AYDEMİR, for his great caring, patience, guidance, and providing me with an exceptional medium for doing research and his expertise, understanding, and patience, added substantially to my master experience. I appreciate his massive knowledge and talent in many fields. He provided me with technical support, direction and became more of a mentor and friend, than a professor. His understanding, determination and politeness lead to easy completing of my master. He is the one professor/teacher who actually made a difference in my life. I doubt that I will ever be able to express my gratefulness fully, but I am indebted him my everlasting appreciation, without whose inspiration and enthusiasm I would not have considered a master career in chemistry research. Furthermore, special thanks go out to Prof. Dr. Akın BAYSAL“and Assoc. Prof. Dr.” Feyyaz DURAP, for their valuable suggestions.
I would also like to acknowledge my family for the support they introduced me through my whole life. I must also thank Dr. Nermin MERİÇ and Dr. Cezmi KAYAN without whose editing assistance and encouragement I would not have completed this study.
In addition,”I would like to thank Prof. Dr.”Yilmaz TURGUT and Prof Dr. Mehmet KARAKAPLAN to offer me starting materials in my study.
In conclusion, this research would not have been possible without the financial support of Dicle University“Science and Technology Application and Research Center”(Project no: 113Z297)“and Technological Research Council of Turkey.”I would like to express my gratitude to those institutions.
II CONTENTS Contents Page No “ACKNOWLEDGEMENTS”... I CONTENTS ... II ABSTRACT” ... IV TABLE LIST ... VI FIGURE LIST ... VII APPENDICES ... VIII SYMBOLS AND ABBREVIATIONS ... IX
1. INTRODUCTION ... 1 2. LITERATURE SURVEY ... 3 2.1. Ferrocene ... 3 2.2. Ferrocenylphosphine Ligands ... 3 2.3. Phosphorus Based-Ligands ... 4 2.4. Phosphinites ... 4 2.5. Transfer Hydrogenation ... 5 3. PREVIOUS STUDIES ... 7
4. MATERIALS and METHODS ... 11
4.1. Chemicals ... 11
4.2. Instrument Used for Characterization ... 12
4.3. Method ... 12
4.4. Synthesis of ferrocene carboxaldehyde (Jia et al. 2011) ... 13
4.5. Synthesis of amino alcohols based on the ferrocene backbone (Ak et al. 2013) ... 14
4.5.1. (2R)-2-[(Ferrocenylmethyl)amino]-2-phenylethan-1-ol ... 15
“ ... 15
4.5.2. (2S)-2-[(Ferrocenylmethyl)amino]-2-phenylethan-1-ol” ... 16
“4.5.3. (2R)-2-[(Ferrocenylmethyl) amino]-3-phenylpropan-1-ol” ... 17
“4.5.4. (2S)-2-[(Ferrocenylmethyl) amino]-3-phenylpropan-1-ol”... 18
4.6. Synthesis of Precursor Alcohols ... 19
4.6.1. N-benzyl-N-[(S)-1-[α-naphthylethyl]amine ... 19
“4.6.2. (1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenyl ethan-1-ol” .... 20
4.6.3. (2S)-1-{benzyl [(1S)-1-(naphthalen-1-yl)ethyl]amino” 3-phenoxypropan-2-ol ... 21
4.7.Synthesis of Phosphinite Ligands Synthesis of phosphinite ligands based on the ferrocene backboneii ... 22
III 4.7.1. (2R)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinite ... 22 4.7.2.(2S)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinite (2) ... 23 4.7.3. (2R)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinite ... 24 4.7.4. (2S)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinite ... 25 4.7.5. (1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyl diphenyl phosphinite (5) ... 26
4.7.6. (2S)-1-{benzyl [(1S)-1-(naphthalen-1-yl)ethyl]amino 3-phenoxypropan-2-yl diphenylphosphinite, (6) ... 27
4.8.Synthesis of The Complexes Synthesis of ferrocene based Ir(III)-phosphinites complexes ... 28
4.8.1.[(2R)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (1a) ... 28
4.8.2. [(2S)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (2a) ... 29
4.8.3. [(2R)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (3a) ... 30
4.8.4.[(2S)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (4a) ... 31
4.8.5.(1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyl diphenylphosphinito[dichloro(η6-p-cymene)ruthenium(II)] (5a) ... 32
4.8.6.(1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyl diphenylphosphinito[dichloro(η6-benzene)ruthenium (II)] (5b) ... 33
4.8.8.(2S)-1-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-3-phenoxypropan-2-yl diphenylphosphinito[dichloro(η6-benzene)ruthenium (II)] (6b) ... 35
4.9.Catalytic Studies ... 36
5. RESULTS and DISCUSSION ... 41
REFERENCES ... 47
SPECTRA ... 53
IV ABSTRACT”
“Asymmetric Transfer Hydrogenation of Ketones with”Ru (II) and Ir (III) Phosphinite complexes
M.Sc. THESIS
Ahmed SABRI
UNIVERSITY OF DICLE
INSTITUTE OF NATURAL AND APPLIED SCIENCES DEPARTMENT OF CHEMİSTRY
2016
Transition metal catalyzed homogeneous asymmetric catalysis is an important field of research, and the development of effective ligands for this process have been withdrawing considerable interest. It is known that the stereoselectivity and reactivity of an enantioselective conversion depend on the structure of the chiral ligand bound to the transition metal, thus, the preparation and design“of efficient chiral ligands are important in this”field. Furthermore, transfer hydrogenation reactions are simple, environmentally benign and easy to handle and reaction conditions are not so harsh.
Since the ferrocene moiety is easily modifiable and has highly electron donating property, it has been widely studied as a backbone of chiral phosphine ligand. Furthermore, ferrocene based ligands usually crystallize easily and they are more stable at ambient condition than similar nonferrocenyl ligands. These properties are useful in their use and purification processes. Moreover, ferrocenes have a unique structure allowing them to develop various“ferrocenyl phosphine ligands, which are”extensively used“in asymmetric reactions.”
In the present work, six precursors for alcohols were prepared, from which six different alcohols were obtained. Then, six phosphinite ligands, 1-6 were synthesized by the reaction of these compounds with Ph2PCl. Treatment of 1-4 with Ir gave Ir(III)
V
complexes, 1a-4a, while reaction of ligands 5 and 6 with [Ru(η6-p-cymene)Cl2]2 or
[Ru(η6
-benzene)Cl2]2 afforded new Ru(II) complexes 5a-b and 6a-b, which were
characterized by spectroscopic methods, such as NMR and IR, and elemental analysis. Finally, these complexes were employed as catalyst in transfer hydrogenation reaction of ketones. Generally, high conversions and with some complexes enantiomeric excess (ee) up to 98 % were obtained.
Key Words: Phosphinite, Ferrocene, Ruthenium, Iridium, Asymmetric Transfer Hydrogenation, Catalysis.
VI
TABLE LIST
Table No Page No
“Table 1. Asymmetric transfer hydrogenation of acetophenone with iso-PrOH
catalysed by complexes 1a-4a.” 36
Table 2. Asymmetric transfer hydrogenation results for substituted
acetophenones catalyzed by complexes 1a-4a. a 37
“Table 3. Asymmetric transfer hydrogenation of acetophenone with iso-PrOH
catalysed by complexes 5a-6b.” 38
“Table 4. Asymmetric transfer hydrogenation results for substituted
acetophenones catalyzed by complexes 5a,5b,6a,6b.a” 39
“Table 5. Asymmetric transfer hydrogenation of various simple ketones with
VII
FIGURE LIST
Figure No Page No Figure 1. Structures of several phosphorus based chiral ligands used in
asymmetric transfer hydrogenation. ... 2
Figure 2. Synthesis of ferrocene ... 3
Figure 3. Use of 2-propanol as a hydrogen source ... 5
Figure 4. Use of HCOOH/Et3N mixture as a hydrogen source ... 5
Figure 5. Asymmetric hydrogenation of α-(N- acetamido)acrylate” ... 7
Figure 6. “Asymmetric transfer hydrogenation of ketones by Ir(I) catalyst” ... 8
Figure 7. Unsymmetrical ferrocenyl-phosphinite ligands ... 8
Figure 8. “A variety of phosphinites based on ferrocenyl moiety.” ... 9
Figure 9. New ferrocenyl phosphinites. ... 9
Figure 10. A is accurate MS1 spectrum of N-benzyl-N-[(S)-1-[α-naphthylethyl]amine. ... 60
Figure 11. A is accurate MS1 spectrum of (1R)-2-{benzyl[(1S) -1-(naphthalen-1-yl)ethyl]amino}-1-phenylethan-1-ol. ... 61
Figure 12. A is accurate MS1 spectrum of (2S)-1-{benzyl[(1S) -1-(naphthalen-1-yl)ethyl]amino 3-phenoxypropan-2-ol. ... 62
VIII
APPENDICES
Spectra No Page No Spectrum 1. 31P-{1H} NMR Spectrum of (2R)-2-(Ferrocenylmethylamino)
-2-phenylethyl diphenylphosphinite (1) ... 53 Spectrum 2. 31P-{1H} NMR Spectrum of (2S)-2-(Ferrocenylmethylamino)
-2-phenylethyl diphenylphosphinite (2) ... 53 Spectrum 3. 31P-{1H} NMR Spectrum of(2R)-2-(Ferrocenylmethylamino)
-3-phenylpropyl diphenylphosphinite (3) ... 54 Spectrum 4. 31P-{1H} NMR Spectrum of (2S)-2-(Ferrocenylmethylamino)
-3-phenylpropyl diphenyl phosphinite (4) ... 54 Spectrum 5. 31P-{1H} NMR spectrum of (1R)-2-{benzyl[(1S)-1-(naphthalen
-1-yl)ethyl] amino}-1-phenylethyldiphenylphosphinite (5) ... 55 Spectrum 6. 31P-{1H} NMR spectrum of (2S)-1-{benzyl [(1S)-1-(naphthalen
-1-yl)ethyl]amino}- 3-phenoxypropan-2-yl diphenylphosphinite,
(6) ... 55 Spectrum 7. 31P-{1H} NMR Spectrum of [(2R)-2-(Ferrocenylmethylamino)
-2-phenylethyldiphenylphosphinito
(dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (1a) ... 56 Spectrum 8. 31P-{1H} NMR Spectrum of [(2S)-2-(Ferrocenylmethylamino)
-2-phenylethyldiphenylphosphinito(dichloro(ɳ5-pentamethylcycl
opentadienyl)iridium(III))] (2a) ... 56 Spectrum 9. 31P-{1H} NMR Spectrum of [(2R)-2-(Ferrocenylmethylamino)
-3-phenylpropyldiphenylphosphinito(dichloro(ɳ5-pentamethylcy
clopentadienyl)iridium(III))] (3a) ... 57 Spectrum 10. 31P-{1H} NMR Spectrum of [(2S)-2-(Ferrocenylmethylamino)
-3-phenylpropyldiphenylphosphinito(dichloro(ɳ5-pentamethylcy
clopentadienyl)iridium(III))] (4a) ... 57 Spectrum 11. 31P-{1H} NMR spectrum of (1R)-2-{benzyl[(1S)-1-(naphthalen-
1-yl)ethyl] amino}-1-phenylethyldiphenylphosphinito
[dichloro(η6-p-cymene)ruthenium(II)] (5a) ... 58 Spectrum 12. 31P-{1H} NMR spectrum of (1R)-2-{benzyl[(1S)-1-(naphthalen-
1-yl) ethyl] ... 58 Spectrum 13. 31P-{1H} NMR spectrum of (2S)-1-{benzyl[(1S)-1-(naphthalen-
1-yl)ethyl]amino}-3-phenoxypropan-2-yldiphenylphosphinito
[dichloro(η6-p-cymene)Ruthenium ... 59 Spectrum 14. 31P-{1H} NMR spectrum (2S)-1-{benzyl[(1S)-1-(naphthalen-
1-yl)ethyl]amino}-3-phenoxypropan-2-yldiphenylphosphinito[di
IX
SYMBOLS AND ABBREVIATIONS
Ar Aryl
ATH Asymmetric Transfer Hydrogenation
CDCl3 Chloroform-d1
CH2Cl2 Dichloromethane
Cod 1,5-cyclooctadiene
Cy2PCl Monochlorodicyclohexylphosphine
min Minute
DEPT Distortionless Enhancement by Polarization Transfer
DMF N,N'-Dimetylformamide
DMSO-d6 Dimethyl sulfoxide-d6
Con. Conversion
ee Enantiomeric excess
Et3N Triethylamine
GC Gas Chromatography
HETCOR Heteronuclear correlation (13C-1H)
IR Infrared
J Coupling constant
NMR Nuclear Magnetic Resonance
Ph2PCl Monochlorodiphenylphosphine
ppm Part Per Million
R Alkyl TH Transfer Hydrogenation THF Tetrahydrofuran h Hour ʋ Frequency (cm-1) δ Chemical shift
1 1. INTRODUCTION
Transition metal catalyzed asymmetric reactions are widely used to synthesize a great deal of enantiomerically pure drugs and chiral compounds. Preparation of effective chiral ligands is very important in this field. Therefore they have appealed considerable interest from both industry and academia, because the stereoselectivity and reactivity of an asymmetric conversion greatly depend on the structure of the chiral ligand (Ghent et al. 2007).
It was seen that chiral Ir and Rh transition metals in reaction medium catalyzed reduction and were effective for a variety of substrates in Hydrogen transfer reactions (Abdur-Rashid et al. 2001).
“Asymmetric hydrogenation is one of the most valuable reactions,”investigated in innumerable studies. Since its early years, the reaction has been used for industrial applications, thanks to its prominent catalytic activity and chiral recognition ability.“Asymmetric hydrogenation is usually promoted by a complex carrying Rh, Ru, or Ir”(Shimuzu et al. 2005).
Ferrocene ligands have gained much attention due to their peculiar chemical features, namely diastereoselective metallation on the cyclopentadienyl ring and retentive nucleophilic displacement on the benzylic position, which allow the preparation of a broad range of substituted derivatives. Numerous ferrocenyl ligands incorporating both planar and central chirality have proved very effective in several asymmetric catalytic processes (Patti and Nicalausi 2000).
Recently, there has been a resurgence of interest in the chemistry of ferrocene and its derivatives because of their increasing applications, for example, as biologically active compounds, in many fields of chemistry including material science and asymmetric catalysis. The synthesis of aryl-substituted ferrocenes has attracted considerable attention because of their use in the design of liquid crystal materials (Long et al. 2010).
The ridged planar chirality enforced by the ferrocene backbone of ferrocenylphosphine is considered to be important to the effectiveness of the catalyst (Ghent et al. 2007).
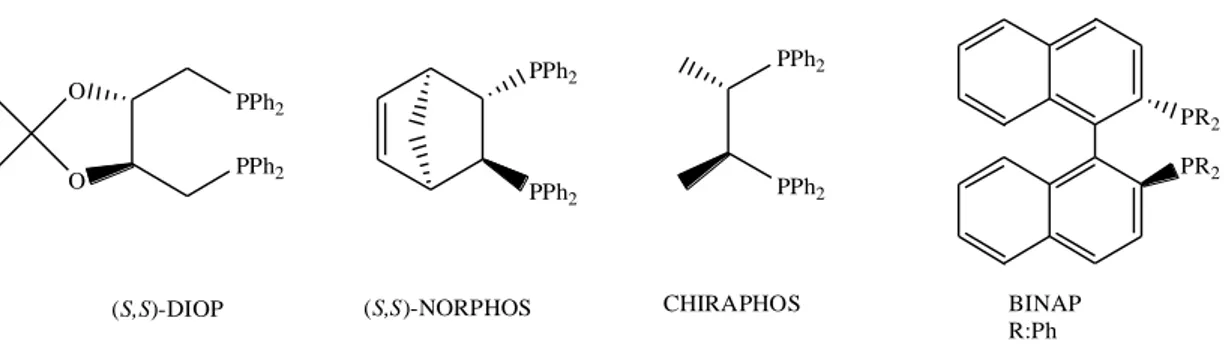
1. INTRODUCTION 2 PPh2 PPh2 (S,S)-NORPHOS PR2 PR2 BINAP R:Ph PPh2 PPh2 O O (S,S)-DIOP PPh2 PPh2 CHIRAPHOS
Figure 1. Structures of several phosphorus based chiral ligands used in asymmetric transfer hydrogenation.
3 2. LITERATURE SURVEY
2.1. Ferrocene
Ferrocene has a formula of Fe (C5H5)2 and it consists“of two cyclopentadienyl
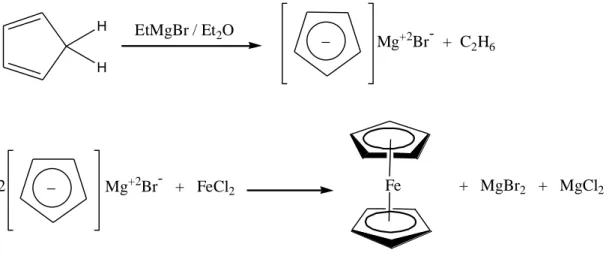
rings bound on opposite sides of a central metal atom.”These type of structure is known as sandwich compounds. The discovery of ferrocene and its a number of analogues made a significant contribution to the rapid development of organometallic chemistry. Actually, ferrocene was first synthesized accidentally. Pauson and Kealy at Duquesne University reacted cyclopentadienyl magnesium bromide with ferric chloride to obtain fulvalene in 1951. However, they got a light orange powder of "astonishing stability" (Figure 1), (Pauson et al. 1951).
H H EtMgBr / Et2O _ Mg+2Br- + C2H6 _ Mg+2Br- + FeCl 2 2 Fe + MgBr2 + MgCl2
Figure 2. Synthesis of ferrocene 2.2. Ferrocenylphosphine Ligands
In recent years, chiral ferrocenylphosphine ligands have attracted considerable attentions due to its great success in catalytic asymmetric reactions. Because they are very efficient for a variety of reactions, such as, allylic alkylation, hydrosilylation, cyclopropanation, Grignard cross-coupling reactions and hydrogenation (Ak et al. 2014). It is well-known that ferrocene and its derivatives are very important compounds for photochemistry and modern organometallic chemistry because of their outstanding stabilities, generating novel materials possessing interesting chemical, electrical, optical, and magnetic properties (Asmafiliz et al. 2009).
2. LITERATURE SURVEY
4 2.3. Phosphorus Based-Ligands
The study of ligands containing phosphorus atoms has been of great interest throughout inorganic and organic chemistry. These ligands have been investigated for the last several decades and have been found to be of highly attention because of their many applications in organometallic chemistry (Zuburi and Woollins 2003).
The coordination and organometallic chemistry of phosphorus bearing ligands possessing one (or more) P-N bond(s) has received some attention especially of late (Rudd et al. 2004).
Although poorly exploited in catalysis, with respect to tertiary phosphines and phosphites, phosphorus(V) derivatives or complexes (generated in situ or performed) stabilised by such ligands are effective in several catalytic reactions. Asymmetric catalysis with chiral variants incorporating an aminophosphine functionality have also been described (Gaw et al. 2002).
Phosphinites are one of the most important phosphorous containing ligands in organometallic chemistry and they have various electronic and steric features. Thus, they are widely used in transition metal catalyzed asymmetric conversions.
2.4. Phosphinites
Phosphinites have been explored extensively and they possess different structural, electronic and chemical features than phosphines. Therefore, they open various prospects to prepare new improved ligands for asymmetric catalysis. For example, M-P bond are usually stronger for phosphinites than the similar phosphines owing to the existence of the electron-withdrawing P-OR group. Furthermore, the valence σ*-orbital of the phosphinite P (OR) R2 is more stable, which rendering it a better acceptor (Galka and
Kraatz 2003).
Despite the proven efficiency of catalysts derived from these ligands, the possibility of discovering improved utility, activity, and selectivity by the design of new ligand classes has continued to stimulate research. Recently, bis (phosphinites), and mixed donor ligands such as (P, O), (P, N), and (P, S) compounds have been of great interest and have proved advantageous in some cases. The last class is by far the least explored and has been used with limited success in catalysis (Hauptman et al. 1998).
5
The introduction of substituents can modify the electron density of the aromatic rings, and this, in turn, alters the electron density at phosphorus. Catalysts prepared from these modified systems can have different reaction rates and selectivities when compared to the parent ligand system (Gergely et al. 2003).
2.5. Transfer Hydrogenation
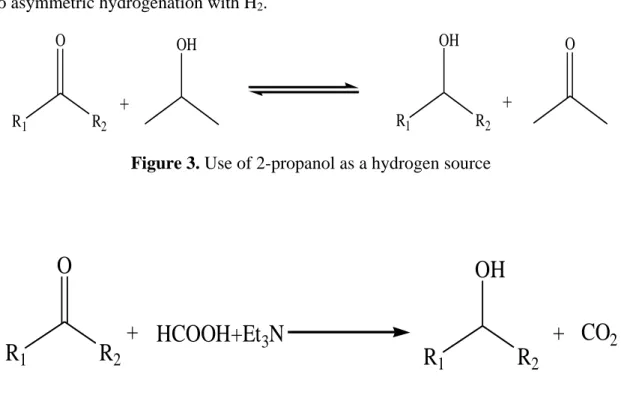
Using a hydrogen donor is more advantageous than that of molecular hydrogen because it eludes the limitations and the risks related“with hydrogen gas as well as the need for pressure vessels and other equipment.”
Transition-metal catalyzed transfer hydrogenation using 2-propanol as a hydrogen source has become an efficient method in organic synthesis as illustrated by several useful applications reported in recent years. The reaction conditions for this important process are economic, relatively mild and environmentally friendly. The most frequently employed catalysts for this reaction are ruthenium (II) complexes; however some rhodium and iridium complexes have been employed as well.”
In view of the low cost of the reducing agent and its operational simplicity,“transition-metal-catalyzed transfer hydrogenation either with 2-propanol”or with an HCO2H/Et3N mixture as a hydride source has appeared as an attractive alternative
to asymmetric hydrogenation with H2.
R1 R2 O OH
+
R1 R2 OH O+
Figure 3. Use of 2-propanol as a hydrogen source
R
1R
2O
+
R
1R
2OH
+
HCOOH+Et
3N
CO
22. LITERATURE SURVEY
7 3. PREVIOUS STUDIES
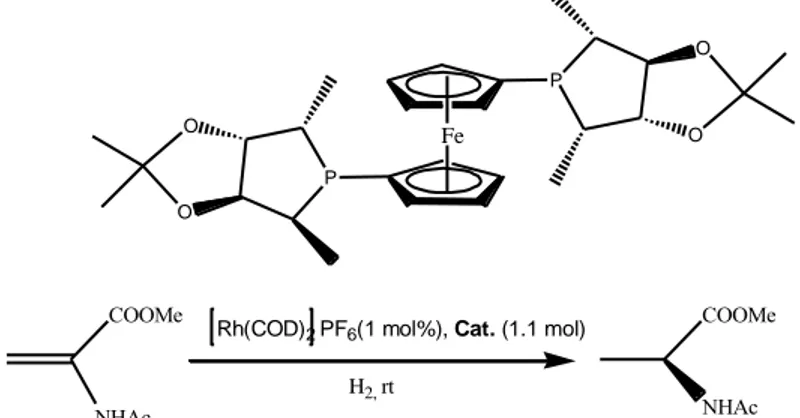
Liu et al. (2002) reported the synthesis of "a new chiral ferrocenyl diphosphine ligand from readily available D-mannitol.”They reported“its Rh-complex exhibited“high enantioselectivity and reactivity in the asymmetric hydrogenation of dehydroamino acid derivatives and itaconic acid derivatives.”They prepared“the catalyst in situ by mixing [Rh(COD)2]-PF6 and 1 in a solvent.”
P O O P O O Fe NHAc COOMe
Rh(COD)2 PF6(1 mol%), Cat. (1.1 mol)
H2, rt
COOMe NHAc
Figure 5. Asymmetric hydrogenation of α-(N- acetamido)acrylate”
Zhang et al. (2008) “synthesized novel planar chiral diferrocenyl phosphinediimines for the iridium catalyzed asymmetric transfer hydrogenation of aromatic ketones. They generated the chiral Ir catalytic system in situ and applied it to asymmetric transfer hydrogenation of aromatic ketones using 2-propanol as a source of hydrogen. The results showed that the corresponding chiral alcohols could be obtained with moderate activity and low enantioselectivities under mild conditions. 4-benzoylpridine was a preferred substrate with respect to catalytic activity in the presence of base.”
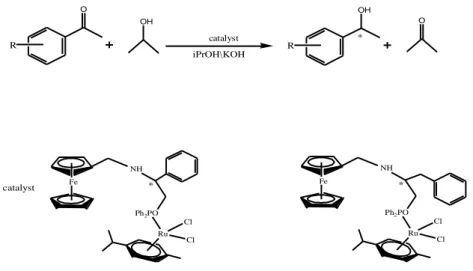
3. PREVIOUS STUDIES 8 Fe PPh2 C CH3 H N H C R HC N C H H3C Fe Ph2P Ph O R Ph R OH Ir(COD)Cl 2/ 3 or 4, iPrOH KOH,reflux a: R= CH3 b: R= CH2CH3 c: R= d: R= N N
Figure 6. “Asymmetric transfer hydrogenation of ketones by Ir(I) catalyst” Aydemir et al. (2011) prepared“phosphinite based chiral Ru(II) complexes (5 and 6) and employed them as catalysts in the asymmetric transfer hydrogenation of acetophenone derivatives.Under optimized conditions, aromatic ketones were reduced in good conversions and in moderate to good enantioselectivities (up to 85% ee).”
Ak et al. (2013) synthesized“unsymmetrical ferrocenyl-phosphinite ligands possessing a stereogenic center. They reported that these ligands“exhibited good enantioselectivities in the ruthenium-catalyzed asymmetric transfer hydrogenation of acetophenone derivatives, and obtained up to 91% ee.”
Fe NH Ph2PO Ru Cl Cl * Fe NH Ph2PO Ru Cl Cl * catalyst R O OH R OH O catalyst iPrOH\KOH *
Figure 7.Unsymmetrical ferrocenyl-phosphinite ligands
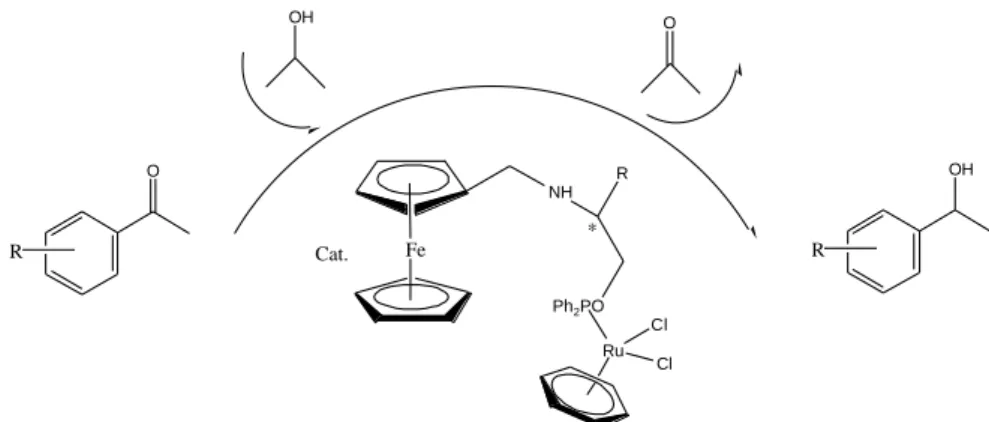
Işık et al. (2013) reported“new examples of enantiomerically pure monodendate phosphinite ligands involving both a ferrocene moiety and NH bridging moiety adjacent to the stereocenter, as well as their ruthenium(II) dichloro complexes”and they“screened the phosphinites based on ferrocenyl moiety possessing stereogenic center“as ligands for
9
ruthenium(II)-catalyzed transfer hydrogenation of aromatic ketones”to afford corresponding secondary alcohols using iso-PrOH as the hydrogen source in the presence of NaOH. They reported that“up to 99% conversion with 97% ee was obtained in the transfer hydrogenation of acetophenone derivatives.”
Fe NH Ph2PO Ru Cl Cl * R Cat. R O OH R OH O
Figure 8. “A variety of phosphinites based on ferrocenyl moiety.”
Ak et al. (2014) “screened a variety of phosphinite based on ferrocenyl moiety possessing central chirality as ligands for ruthenium(II)-catalyzed transfer hydrogenation of acetophenone derivatives using iso-PrOH as the hydrogen source to afford the corresponding product, (R) or (S)-1-phenylethanol derivatives with high conversions and good enantioselectivities.” They “employed these complexes in the asymmetric reduction of different prochiral ketones (up to 85% ee).””
Fe NH Ph2PO Ru Cl Cl * R R: D-isopropyl 9; L-isopropyl 10 R: D-isobutyl 11; L-secbutyl 12
3. PREVIOUS STUDIES
11 4. MATERIALS and METHODS 4.1. Chemicals
1. Ferrocene 21. (R)-Styrene oxide 41. 2-Propanol 2. Triethylorthoformate 22. (S)-glycidyl phenyl
ether
42. Acetophenone
3. Aluminiumchloride 23. n-Hexane 43. 4-Fluoroacetophenone 4. Sodium hydrosulfite 24. Tetrahydrofuran 44. 4-Bromoacetophenone 5. D-Phenylglycinol 25. Diethylether 45. 4-Chloroacetophenone 6. L-Phenylglycinol 26. Petroleum ether 46. 4-Nitroacetophenone 7. D-Phenylalaninol 27. Ethyl acetate 47. 2-Methoxyacetophenone 8. L-Phenylalaninol 28. Toluene 48. 3-Methoxyacetophenone 9. Chlorodiphenylphosphine 29. Chloroform 49. 4-Methoxyacetophenone 10. Triethylamine 30. Dichloromethane 50. 3-Nitroacetophenone
11. [Ir(η5-C5Me5)(μ-Cl)Cl]2 31. Methanol 51. 2′-(Trifluoromethyl)acetophenone
12. [Ru(η6-p-cymene)(µ-Cl)Cl]2, 32. Sodium 52. 3′-(Trifluoromethyl)acetophenone
13. [Ru(η6-benzene)(µ-Cl)Cl]2 33. Calcium hydride 53. 4′-(Trifluoromethyl)acetophenone
14. (S)-(-)-1-[Naphthalen-1-yl-ethy ]amine
34. di-fosforpentaoksit 54. 2-Methylacetophenone
15. Chloroform-d 35. Sodium hydroxide 55. 3-Methylacetophenone 16. Sodium borohydride 36. Benzaldehyde 56. 4-Methylacetophenone 17. Potassium hydroxide 37. Benzophenone 57. Ethyl methyl ketone 18. Potassium carbonate 38. Acetone 58. Methyl 1-naphthyl ketone 19. Butyl methyl ketone 39. İsopropylmethyl
ketone
59. Isobutyl methyl ketone
4. MATERIALS and METHODS
12 4.2. Instrument Used for Characterization 1. FTIR Spectrometer“(Mattson 1000 ATI UNICAM)” 2. Elemental analysis“(Fisons EA 1108 CHNS-O)” 3. NMR Spectrometer (Bruker AV400)
4. Gas chromatograph (Shimadzu GC 2010 Plus) 5. Melting points (Gallenkamp MPD 350 BM 2.5) 6. Polarimeter (Perkin Elmer 341)
4.3. Method
The study can be outlined with three main titles: i. Synthesis of precursor alcohols
ii. Synthesis of ferrocene based C2-symmetric bis(phosphinite) ligands,
iii. Synthesis of bis(phosphinite) Ru(II)-benzene complexes,
iv. Application of bis(phosphinite)-Ru(II)-benzene complexes as catalyst“in transfer hydrogenation reactions”and determining their catalytic activity. All experimental studies, i.e. synthesis of iso-propyl based C2-symmetric
bis(phosphinites) and their Ru(II)benzene complexes, and use of them in catalytic investigations were accomplished according to the literature (Ak et al. 2015).
13
4.4. Synthesis of ferrocene carboxaldehyde (Jia et al. 2011)
To a three-neck flask were added 10.0 g of dry ferrocene (53.76 mmol) and 150 mL of CH2Cl2. Next 39.2 g of triethyl orthoformate (264.34) was added dropwise to the
mixture with stirring. After the ferrocene was completely dissolved, 30.0 g of anhydrous AlCl3 was added slowly, and the reaction mixture was stirred at room temperature for 4 h.
The reaction was quenched with sodium hydrosulphite saturated solution (200 mL) and the mixture was extracted with diethyl ether (200 mL). After being concentrated under reduced pressure, the residue was purified by chromatography on silica gel (petroleum ether:ethyl acetate = 5:1) to afford a red solid (7 g) with a yield of 70%.
Fe CHO Fe + CH(OC2H5)3 + AlCl3 CH2Cl2 1 H NMR (CDCl3, ppm): δ 4.28 (s, 5H, C5H5), 4.61 (s, 2H, C5H4); 4.80 (s, 2H, C5H4), 9.96 (s, 1H, CHO).
4. MATERIALS and METHODS
14
4.5. Synthesis of amino alcohols based on the ferrocene backbone (Ak et al. 2013)
Ferrocenecarboxaldehyde (4.0 mmol) and D-phenyl glycinol (4.2 mmol) were dissolved in previously dried chloroform (40 mL; dried over K2CO3) and the resulting
solution was heated at reflux under argon for 90 min. Next, the solution was allowed to cool to room temperature, the solvent was removed under reduced pressure and the red– brown residue immediately redissolved in dry methanol (40 mL; distilled from a MeONa solution). The methanolic solution was cooled in an ice bath and treated slowly with solid NaBH4 (20 mmol over 30 min). After adding all of the NaBH4, the mixture was stirred at o
C for 1 h and at room temperature for a further 90 min. Next, the cooled mixture was quenched with an aqueous solution of NaOH (10%, 40 mL) and extracted with CH2Cl2 (2
x 40 mL). The combined organic layers were washed with brine (2 x 40 mL), dried over anhydrous magnesium sulfate, and evaporated, leaving the crude product as a yellow– brown solid. Subsequent purification of the residue by column chromatography (silica gel, dichloromethane–methanol 10:1) led to the development of two bands: the first (minor) one containing mostly ferrocenyl methanol, followed by the major band of the amino alcohol. Careful evaporation of the second fraction afforded pure product as an amber oil, which slowly solidified to a brown solid.
15
4.5.1. (2R)-2-[(Ferrocenylmethyl)amino]-2-phenylethan-1-ol
Yield: 1.03 g, 77 %; mp: 78–79 oC; [α]D20= - 42.8o“(c 1.2, MeOH); 1H NMR
(CDCl3, ppm): δ 2.10 (br, 2H, NH and OH), 3.29 (d, 1H, J = 12.6 Hz, CH2NH, (a)), 3.43
(d, 1H, J = 12.6 Hz, CH2NH, (b)), 3.48– 3.53 (m, 1H, CH2OH) (a)), 3.63–3.66 (m,
1H,”CH2OH) (b)), 3.75– 3.79 (m, 1H, –CHNH), 4.01 (m, 5H, C5H5+2H, C5H4), 4.06 (m,
2H, C5H4), 7.12–7.33 (m, 5H, C6H5); 13C NMR (CDCl3, ppm): δ 46.29 (CH2NH), 63.98
(CHNH), 66.58 (CH2OH), 67.79, 67.89, 68.09, 68.42 (C5H4), 68.49 (C5H5), 85.52
(i-C5H4), 127.32, 127.75, 128.73 (C6H5), 140.37 (i-C6H5); “IR (KBr pellet in cm-1) ν: (N–
H): 3281, (C-Cp): 3086, (C=C-Cp): 1455, (O–H): 3280; Anal. Calcd for C19H21NOFe
(335.27 g/mol): C 67.87; N 4.16; H 6.29. Found: C 67.82; N 4.11; H 6.24.” “ Ph OH NH2 H + NaBH4 Fe CHO Fe NH Ph HO
4. MATERIALS and METHODS
16
4.5.2. (2S)-2-[(Ferrocenylmethyl)amino]-2-phenylethan-1-ol”
Yield: 0.98 g, 73%; mp: 78–79 oC; [α]D20= +42.8o“(c 1.2, MeOH); 1H NMR
(CDCl3, ppm):”δ 2.34 (br, 2H, NH and OH), 3.39 (d,1H, J = 13.2 Hz, CH2NH, (a)),
3.51–3.63 (m, 2H, CH2NH (b) + CH2OH(a)), 3.74 (dd, 1H, J = 4.2 and 11.0 Hz CH2OH)
(b)), 3.87 (dd, 1H, J = 4.4 and 8.8 Hz, –CHNH), 4.10 (m, 5H, C5H5 + 2H, C5H4), 4.18
(m,2H, C5H4), 7.35–7.43 (m, 5H, C6H5); 13C NMR (CDCl3, ppm): δ 46.31 (CH2NH),
63.98 (CHNH), 66.64 (CH2OH), 67.80,67.90, 68.11, 68.43 (C5H4), 68.51 (C5H5), 86.45
(i-C5H4), 127.34,127.72, 128.73 (C6H5), 140.37 (i-C6H5); “IR (KBr pellet in cm-1) ν :(N–
H): 3291, (C-Cp): 3087, (C=C-Cp): 1441, (O–H): 3320; Anal.Calcd for C19H21NOFe
(335.27 g/mol): C 67.87; N 4.16; H 6.29.Found: C 67.81; N 4.10; H 6.22.” Ph OH NH2 H + NaBH4 Fe CHO Fe NH Ph HO
17 “4.5.3. (2R)-2-[(Ferrocenylmethyl) amino]-3-phenylpropan-1-ol” Yield:1.19 g, 81%; mp: 50–51 oC; [α]D20= +18.6o“(c 1.2, MeOH); 1H NMR (CDCl3, ppm): δ 2.11 (br, 2H, NH and OH), 2.83–2.88(m,”2H, CH2Ph), 3.00–3.06 (m, 1H, CHNH), 3.39–3.45 (m, 2H, CH2NH (a) + CH2OH (a)), 3.54 (d, 1H, J = 13.2 Hz, CH2NH (b)), 3.70 (dd, 1H, J = 3.8 and 10.6, CH2OH (b)), 4.09 (m, 5H, C5H5 + 2H, C5H4), 4.16 (m, 2H, C5H4), 7.23–7.36 (m, 5H, C6H5); 13C NMR (CDCl3, ppm): δ 38.26 (CH2Ph) 46.07 (CH2NH), 59.71 (CHNH), 62.38 (CH2- OH), 67.61, 67.64, 67.76, 67.86 (C5H4), 68.35 (C5H5), 86.85 (i-C5H4) 126.62, 128.72, 129.20 (C6H5),138.52 (i-C6H5); “IR (KBr pellet in cm-1 ) ν: (N–H): 3268, (C-Cp): 3084, (C=C-Cp): 1448;(O–H): 3305; Anal. Calcd for C20H23NOFe (349.29 g/mol): C 68.76; N 4.02; H 6.64. Found: C 68.74; N
3.99; H 6.59.” OH NH2 H + Ph NaBH4 Fe CHO Fe NH HO Ph
4. MATERIALS and METHODS 18 “4.5.4. (2S)-2-[(Ferrocenylmethyl) amino]-3-phenylpropan-1-ol” Yield: 1.20 g, 82%; mp: 50–51 oC; [α]D20 = -18.6o“(c 1.2, MeOH); 1H NMR (CDCl3, ppm): δ 2.01 (br, 2H, NH and OH), 2.82–2.85 (m,”2H, CH2Ph), 3.01–3.05 (m, 1H, CHNH), 3.40 (m, 1H, CH2OH (a), 3.43 (d, 1H, J = 13.1 Hz, CH2NH (a), 3.54 (d, 1H, J = 12.9 Hz, CH2NH (b)), 3.70 (dd, 1H, CH2OH (b)), 4.10 (m,5H, C5H5 + 2H, C5H4), 4.16 (br s, 2H, C5H4), 7.23–7.36 (m, 5H,C6H5); 13C NMR (CDCl3, ppm): δ 38.28 (CH2Ph), 46.05 (CH2NH), 59.68 (CHNH), 62.38 (CH2OH), 67.60, 67.63, 67.76, 67.85 (C5H4), 68.35 (C5H5), 86.88 (i-C5H4), 126.62, 128.72, 129.20 (C6H5), 138.52
(i-C6H5);“IR (KBr pellet in cm-1) ν: (N–H): 3268, (C-Cp): 3089, (C=C-Cp): 1448;”(O–
H): 3305; Anal. Calcd for C20H23NOFe (349.29 g/mol): C 68.76; N 4.02; H 6.64. Found:
C 68.73; N 4.00; H 6.58. OH NH2 H + Ph NaBH4 Fe CHO Fe NH HO Ph
19 4.6. Synthesis of Precursor Alcohols
4.6.1. N-benzyl-N-[(S)-1-[α-naphthylethyl]amine NH Ph NH2 1.PhCHO/CH3OH 2. NaBH4 * *
(S)-(-)-1-[Naphthalen-1-yl-ethy]amine (1.00 g, 5.84 mmol) was dissolved in anhydrous MeOH (5 mL) and heated to reflux. Benzaldehyde (0.62 g, 5.84 mmol) was added dropwise over a period of 2 min and the mixture stirred at reflux for 3 h. The solution was allowed to cool to room temperature and sodium borohydride (0.24 g, 6.13 mmol) was added portionwise. After the vigorous effervescence had subsided the mixture was mixtured for 2 h and then heated to reflux for 30 min. The reaction was then quenched by the addition of water (5 mL) and the aqueous phase extracted with DCM (3×10 mL). The separated organics were dried over MgSO4 and filtered. The solution was
concentrated under reduced pressure. The residue was purified by silica-gel column chromatography (n-hexane/EtOAc = 2/1), to afford product (1.01 g, 66 %) as an oil. [α]D20 = +21.3o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.59 (d, 3H, J = 6.6 Hz, -CH3),
1.81 (br, 1H, -NH), 3.75-3.86 (m, 2H, -CH2Ph), 4.76 (q, 1H, J = 6.6 Hz -CHCH3),
7.30-7.39 (m, 5H, ArH), 7.54-7.59 (m, 3H, ArH), 7.82-7.84 (m, 2H, ArH), 7.93-7.95 (m, 1H, ArH), 8.22 (d, 1H, J =6.7 Hz, ArH);13C NMR (CDCl3, ppm): δ 23.69 (-CH3), 51.92
(-CH2Ph), 53.06 (-CHCH3), 122.94, 123.04, 125.33, 125.74, 125.79, 126.94, 127.27,
128.24, 128.42, 128.98, 131.41, 134.04, 140.67, 140.99 (ArC); IR(cm-1): υ 3051, 3028 (aromatic C-H), 2964, 2925 (aliphatic C-H) cm-1; Anal. Calc. for C19H19N (261.37
g/mol): C 87.31, H 7.33, N 5.36; found C 87.19, H 7.28, N 5.29. IT-TOF MS ([M+H]+): 262.15 g/mol.
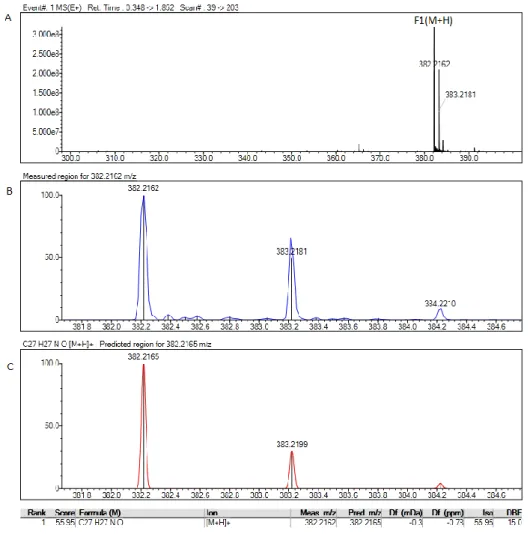
4. MATERIALS and METHODS 20 “4.6.2. (1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenyl ethan-1-ol” NH Ph N Ph Ph HO O Ph MeOH * * *
(R)-Styrene oxide (0.51 g, 4.21 mmol) was added to the solution of
N-benzyl-N-(S)-1-α-naphthylethylamine (1.00 g, 3.83 mmol) in methanol (5 ml). The
solution was heated to 70oC and the reaction was followed by TLC (n-hexane/EtOAc/TEA = 80/10/1). After 24 h the reaction finished, the solvent was removed. The crude product was purified by silica-gel column chromatography ((n-hexane//EtOAc / Et3N = 80/10/1) to afford 1 (1.02 g, 70 %) as a white solid. M.p =
104-106 oC. M.p = 104-106 oC. [α]D20 = +16.4o (c 1, CHCl3). 1H NMR (CDCl3, ppm): δ
1.66 (d, J = 6.8 Hz, 3H, -CH3), 2.79-2.85 (m, 1H, -CH2N, (a)), 2.95-3.00 (m, 1H, -CH2N,
(b)), 3.37 (s, 1H, OH), 3.76 (s, 2H, -CH2Ph), 4.03-4.06 (m, 1H, -CHPh), 4.88 (q, J = 6.8
Hz, 1H, -CHCH3), 7.15-7.30 (m, 10H, Ar-H), 7.48-7.60 (m, 4H, Ar-H), 7.83 (d, J = 8.1
Hz, 1H, Ar-H), 7.90 (d, J = 9.5 Hz, 1H, Ar-H), 8.16 (d, J = 9.3 Hz, 1H, Ar-H); 13C NMR (CDCl3, ppm): δ 14.76 (-CH3), 56.57, 56.59 (-CH2Ph, -CHCH3), 61.31 (-CH2N), 70.99
(-CHPh), 124.03, 124.53, 125.06, 125.70, 125.82, 126.11, 127.30, 127.36, 128.25, 128.55, 128.93, 128.97, 132.25, 134.10, 138.70, 139.92, 142.55 (Ar-C); IR (cm-1): υ 3394 (OH), 3058, 3030 (aromatic C-H), 2965, 2940, 2906 (aliphatic C-H) cm-1; Anal. Calc. for C27H27NO (381.52 g/mol): C 85.00, H 7.13, N 3.67; found C 84.90, H 7.05, N 3.60;
21 4.6.3. (2S)-1-{benzyl [(1S)-1-(naphthalen-1-yl)ethyl]amino”3-phenoxy propan-2-ol NH Ph N Ph HO O MeOH OPh OPh * * *
(S)-glycidyl phenyl ether (0.63 g, 4.21 mmol) was added to the solution of
N-benzyl-N-(S)-1-α-naphthylethylamine (1.00 g, 3.83 mmol) in methanol (5 ml). The
solution was heated to 60oC and the reaction was followed by TLC (n-hexane/EtOAc/TEA = 40/8/3). After 24 h the reaction finished, the solvent was removed. The crude product was purified by silica-gel column chromatography (n-hexane/EtOAc / Et3N = 40/8/3) to afford 2 (1.44 g, 91 %) as a viscous product [α]D20 =
+80.4o (c 1, CHCl3). 1H NMR (CDCl3, ppm): δ 1.68 (d, J = 6.7 Hz, 3H, -CHCH3), 2.68
(s, 1H, OH), 2.82-2.89 (m, AB system, 2H, CH2N), 3.02 (m, 1H, CH2OPh (a)), 3.55 (m,
1H, CH2OPh (b)), 3.73-3.75 (m, 1H, CHCH2),“3.90 (s, 2H, CH2Ph), 4.83 (q, J = 6.7 Hz,
1H, -CHCH3), 6.45 (d, J = 8.1 Hz, 2H, Ar-H), 6.97 (t, J = 7.3 Hz, 1H, Ar-H), 7.22-7.62
(m, 11H, Ar-H), 7.83 (d, J = 8.2 Hz, 1H, Ar-H), 7.93 (d, J = 8.1 Hz, 1H, Ar-H), 7.99 (d, J = 8.6 Hz, 1H, Ar-H);”13C NMR (CDCl3, ppm): δ 12.03 (-CHCH3), 53.29 (-CH2N),
55.23 (-CHCH3), 57.59 (-CH2Ph), 68.02 (-CHCH2), 70.46 (-CH2OPh), 114.35, 120.70,
124.72, 124.83, 125.07, 125.60, 125.88, 127.55, 128.23, 128.50, 128.77, 129.30, 129.58, 132.30, 134.14, 138.72, 139.51, 158.55 (Ar-C); IR (cm-1): υ 3445 (OH), 3050, 3029 (aromatic C-H), 2968, 2923, 2834 (aliphatic C-H) cm-1; Anal. Calc. for C28H29NO2
(411.54 g/mol):“C 81.72, H 7.10, N 3.40; found C 81.66, H 7.01, N 3.32;”IT-TOF MS ([M+H]+): 412.23 g/mol.
4. MATERIALS and METHODS
22
4.7.Synthesis of Phosphinite Ligands Synthesis of phosphinite ligands based on the ferrocene backboneHata! Yer işareti tanımlanmamış.
The alcohol (0.30 mmol) and triethylamine (0.30 mmol) were dissolved in dry toluene (20 mL) under an argon atmosphere. Next PPh2Cl (0.30 mmol) was added
dropwise with a syringe to this solution. The mixture was stirred at room temperature for 30 min. The white precipitate was then filtered under argon and the remaining organic phase was dried in vacuo to produce a white viscous oily compound.
4.7.1. (2R)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinite (1) Fe N H HO Ph + PPh2Cl Toluen -Et3NHCl + Et3N Fe N H O Ph PPh2 Yield: 0.135 g, 87%; [α]D20 = -55.6o (c 1.2, MeOH); 1H NMR (CDCl3, ppm): δ 3.32 (d, 1H, J = 13.2 Hz, CH2NH (a)), 3.50 (d, 1H, J = 13.2 Hz, CH2NH (b)), 3.96–3.99 (m, 2H, CH2OP), 4.12–4.18 (m, 1H, CHNH + 4H C5H4 + 5H, C5H5), 7.32–7.56 (m, 5H,C6H5 + 10H C6H5P); 13C NMR (CDCl3, ppm): δ 46.37 (CH2-NH), 63.52 (d, J = 8.0 Hz, CHNH), 67.59, 67.79, 68.02, 68.24 (C5H4), 68.42 (C5H5), 74.85 (d, J = 17.11 Hz, CH2OP), 87.25 (i-C5H4), 127.67, 127.93, 128.54 (CHC6H5), 128.46 (d, J = 7.0 Hz, m-carbons of phenyls), 129.49 (s, p-carbons of phenyls), 130.59 (d, J = 21.6 Hz,
o-carbons of phenyls), 140.23 (i-C6H5), 141.69 (t, J = 19.31 Hz, i-carbons of phenyls);
31
P-{1H} NMR (CDCl3, ppm): δ 116.30 (s, O-P(Ph)2); IR (KBr pellet in cm-1) ν: (N–H)
= 3323, (C-Cp): 3058, (C=C-Cp): 1454, (P-Ph): 1443, (O–P): 1028; Anal. Calcd for C31H30NOPFe (520.41 g/mol): C 71.54; N 2.69; H 5.81. Found: C 71.52; N 2.67; H 5.79.
23 4.7.2.(2S)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinite (2) Fe N H HO Ph + PPh2Cl Toluen -Et3NHCl + Et3N Fe N H O Ph PPh2 Yield: 0.140 g, 90 %; [α]D20 = +55.6o (c 1.2, MeOH); 1H NMR (CDCl3, ppm): δ 3.30 (d, 1H, J = 13.3 Hz, CH2NH(a)), 3.47 (d, 1H, J = 13.3 Hz, CH2NH (b)), 3.93–3.97 (m, 2H, CH2OP), 4.11–4.16 (m, 1H, CHNH + 4H C5H4 + 5H, C5H5), 7.38–7.55 (m, 5H, C6H5 + 10H C6H5P); 13C NMR (CDCl3, ppm): δ 46.36 (CH2-NH), 63.52 (d, J = 7.10 Hz, CHNH), 67.56, 67.75, 67.98, 68.20 (C5H4), 68.39 (C5H5), 74.85 (d, J = 17.10 Hz, CH2OP), 87.31 (i-C5H4), 127.63, 127.90, 128.50 (CHC6H5), 128.42 (d, J = 6.00 Hz, m-carbons of phenyls), 129.46 (s, p-carbons of phenyls), 130.56 (d, J = 22.1 Hz, o-carbons of phenyls), 140.27 (i-C6H5), 141.67 (t, J = 19.01 Hz, i-carbons of phenyls);
31
P-{1H} NMR (CDCl3, ppm): δ 116.27 (s, O-P(Ph)2); IR (KBr pellet in cm -1
) ν: (N–H) = 3325, (C-Cp): 3060, (C=C-Cp): 1458, (P-Ph): 1440, (O–P): 1030; Anal. Calcd for C31H30-NOPFe (520.41 g/mol): C 71.54; N 2.69; H 5.81. Found: C 71.50; N 2.65; H 5.77.
4. MATERIALS and METHODS 24 4.7.3. (2R)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinite (3) Fe N H HO + PPh2Cl Toluen -Et3NHCl + Et3N Fe N H O PPh2 Ph Ph Yield: 0.140 g, 92%; [α]D20 = +25.4o (c 1.2, MeOH); 1H NMR (CDCl3, ppm): δ 2.84–2.89 (m, 1H, CH2C6H5 (a)), 2.96–3.01 (m, 1H, CH2C6H5 (b)), 3.22–3.25 (m, 1H, CHNH), 3.55 (d, 1H, J = 12.9 Hz, CH2NH (a)), 3.64 (d, 1H, J = 12.9 Hz, CH2NH (b)), 3.92–3.96 (m, 2H, CH2OP), 4.09 (s, 5H, C5H5), 4.14–4.22 (m, 4H, C5H4), 7.25–7.50 (m, 5H, C6H5 + 6H, p and m-C6H5P), 7.60–7.66 (m, 4H, o-C6H5P); 13C NMR (CDCl3, ppm): δ 38.44 (CH2C6H5), 46.73 (CH2NH), 59.88 (d, J = 8.0 Hz, CHNH), 67.65, 67.70, 67.97, 67.98 (C5H4), 68.41 (C5H5), 71.53 (d, J = 17.1 Hz, CH2OP), 87.35 (i-C5H4), 128.34, 128.59,129.15 (CH2C6H5), 129.42 (d, J = 2.2 Hz, m-carbons of phenyls),129.51 (s, p-carbons of phenyls), 130.60 (d, J = 21.6 Hz, o-carbons of phenyls), 137.94 (i-C6H5),
142.08 (d, J = 17.1 Hz, i-carbons of phenyls); 31P-{1H} NMR (CDCl3, ppm): δ 115.62 (s,
O-P(Ph)2); IR (KBr pellet in cm-1) ν: (N–H) = 3322, (C-Cp): 3068, (C=C-Cp): 1454,
(P-Ph): 1435, (O–P): 1026; Anal. Calcd for C32H32-NOPFe (533.43 g/mol): C 72.05; N
25 4.7.4. (2S)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinite (4) Fe N H HO + PPh2Cl Toluen -Et3NHCl + Et3N Fe N H O PPh2 Ph Ph Yield:0.142 g, 93 %; [α]D20 = -25.4o (c 1.2, MeOH); 1H NMR (CDCl3, ppm): δ 2.81–2.86 (m, 1H CH2- C6H5, (a)), 2.89–2.94 (m, 1H, CH2C6H5, (b)), 3.19 (br, 1H, CHNH), 3.52 (d, 1H, J = 12.4 Hz, CH2NH (a)), 3.61 (d, 1H, J = 12.4 Hz, CH2NH (b)), 3.88 (br, 2H, CH2OP), 4.05 (br, 5H, C5H5), 4.10–4.16 (m, 4H, C5H4), 7.19–7.41 (m, 5H, C6H5 + 6H, p- and m-C6H5P), 7.56 (m, 4H, o-C6H5P); 13C NMR (CDCl3, ppm): δ 38.16 (CH2C6- H5), 46.60 (CH2NH), 59.77 (d, J = 7.0 Hz, CHNH), 67.67, 67.72, 68.07, 68.36 (C5H4), 68.46 (C5H5), 71.20 (d, J = 17.1 Hz, CH2OP), 87.25 (i-C5H4), 128.31, 128.40,
128.53 (CH2C6H5), 129.34 (s, p-carbons of phenyls), 129.43 (d, J = 7.0 Hz, m-carbons of
phenyls), 130.54 (d, J = 21.6 Hz, o-carbons of phenyls), 138.72 (i-C6H5), 141.84 (d, J =
18.3 Hz, i-carbons of phenyls); 31P-{1H} NMR (CDCl3, ppm): δ 114.64 (s, O-P(Ph)2);
IR (KBr pellet in cm-1) ν: (N–H) = 3332, (C-Cp): 3054, (C=C-Cp): 1494, (P-Ph): 1435, (O–P): 1023; Anal. Calcd for C32H32NOPFe (533.43 g/mol): C 72.05; N 2.62; H 6.04.
4. MATERIALS and METHODS 26 4.7.5. (1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyl diphenylphosphinite(5) N Ph Ph HO N Ph Ph Ph2PO PPh2Cl CH2Cl2 + Et3NHCl (1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethan-1-ol (0.100 gr, 0.26 mmol) and Et3N (0.027 gr, 0.26 mmol) were dissolved in dry CH2Cl2 (30 mL)
under an argon atmosphere. Next PPh2Cl (0.059 gr, 0.26 mmol) was added dropwise with
a syringe to this solution. The mixture was stirred at 0 oC temperature for 30 minute, and the solvent was removed under reduced pressure. After addition of dry thf, the white precipitate (triethylammonium chloride) was filtered off under argon and dried in vacuo to produce a white viscous oily compound 5. Yield: 0.14 g, 94.7 %. []D20= +34.3 (c 1,
CH2Cl2). 1H NMR (CDCl3, ppm): δ1.66 (d, J = 6.9 Hz, 3H, -CHCH3), 2.83 (m, 1H, -CH2N (a)), 2.98 (m, 1H, -CH2N (b)), 3.77 (s, 2H, -CH2Ph), 4.04-4.06 (m, 1H, CHPh), 4.88 (q, J = 6.9 Hz, 1H, -CHCH3), 6.90 (d, J = 7.6 Hz, 1H, Ar-H), 7.01 (m, 1H, Ar-H ), 7.17-7.92 (m, 24 H, Ar), 8.16 (d, J = 9.1 Hz, 1H, Ar-H); 13C NMR (CDCl3, ppm): δ 14.72 (CHCH3), 56.53, 57.08 (CH2Ph, CHCH3), 61.28 (CH2N), 70.91 (CHPh), 123.96, 124.47, 125.00, 125.75, 126.05, 127.03, 127.69, 128.04, 128.18, 128.48, 128.86, 129.48, 132.02, 134.03, 138.65, 139.87, 142.50 (aromatic carbons), 125.16 (d, J = 4.0 Hz,
m-carbons of OPPh2), 125.63 (s, p-carbons of PPh2), 127.26 (d, J = 6.0 Hz, o-carbons of
OPPh2), 135.38 (d, J = 19.1 Hz, i-carbons of OPPh2); 31P-{1H} NMR (CDCl3, ppm): δ
108.80 (s, O-PPh2); IR (cm-1): υ 3054 (aromatic C-H), 2967 (aliphatic C-H), 1437 (P-Ph),
971 (O-P);Anal. Calc. for C39H36NOP (565.69 g/mol): C 82.81, H 6.41, N 2.48; found C
27 4.7.6. (2S)-1-{benzyl [(1S)-1-(naphthalen-1-yl)ethyl]amino3-phenoxy propan-2-yldiphenylphosphinite, (6) + Et3NHCl N Ph OPh HO N Ph OPh Ph2PO PPh2Cl CH2Cl2
The same protocol for 5 was applied to obtain white viscous oily compound 6 (Yield: 0.14 g, 96.8 %). []D20= +83.0 (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.55 (d, J = 6.6 Hz, 3H, -CHCH3), 2.64 (m, 1H, CH2OPh (a)), 2.79-2.85 (m, AB system, 2H,
CH2N), 2.70-2.85 (m, AB system, 2H, CH2N), 3.35 (m, 1H, CH2OPh (b)), 3.89 (s, 2H,
CH2Ph), 4.10-4.12 (m, 1H, CHCH2), 4.68 (q, J = 6.6 Hz, 1H, CHCH3), 5.90 (d, J = 8.2
Hz, 2H, Ar-H), 6.82 (t, J = 7.3 Hz, 1H, Ar-H), 7.05 (t, J = 7.8 Hz, 2H, Ar-H), 7.23-7.86 (m, 22H, Ar-H); 13C NMR (CDCl3, ppm): δ 10.34 (-CHCH3), 50.89 (d, J = 5.0 Hz,
-CH2N), 53.36 (CHCH3), 57.84 (CH2Ph), 69.66 (-CH2OPh), 78.32 (d, J = 19.1Hz,
-CHCH2), 114.11, 119.99, 124.87, 125.42, 127.55, 128.20, 128.30, 128.78, 128.84,
129.23, 130.07, 130.49, 130.71, 132.13, 133.95, 138.47, 138.76, 158.33 (aromatic carbons), 128.04 (d, J = 7.0 Hz, m-carbons of OPPh2), 128.41 (s, p-carbons of OPPh2),
130.08 (d, J = 21.2 Hz, o-carbons of OPPh2), 142.51 (d, J = 25.2 Hz, i-carbons of OPPh2);
31
P-{1H} NMR (CDCl3, ppm): δ 113.60 (s, O-PPh2); IR (cm-1): υ 3055 (aromatic C-H),
2965 (aliphatic C-H), 1438 (P-Ph), 971 (O-P); Anal. Calc. for C40H38NO2P (595.72
4. MATERIALS and METHODS
28
4.8.Synthesis of The Complexes Synthesis of ferrocene based Ir(III)-phosphinites complexes
At first, [Ir(η5-C5Me5)(μ-Cl)Cl]2 (0.096 mmol) and the phosphinite (0.192 mmol)
were dissolved in 20 mL of toluene and stirred for 1 h at room temperature. The volume was concentrated to ca. 1–2 mL under reduced pressure and addition of n-hexane (15 mL) gave the corresponding complex as an orange microcrystalline solid. The product was collected by filtration and dried in vacuo.
4.8.1.[(2R)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (1a)
Fe N H O Ph PPh2 Fe N H O Ph PPh2 Ir Cl Cl Toluene 1/2[Ir(5-C 5Me5)(-Cl)Cl]2 Yield: 0.150 g, 84.9 %; mp:131-1330C; [α]D20 = -15.8 o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.38 (d, 15H, 4J=2.4 Hz, CH3 of Cp* (C5Me5)), 3.23 (d, J=13.0 Hz, 1H, CH2NH, (a)), 3.43 (d, J=13.1 Hz, 1H, CH2NH, (b)), 3.79 (br, 1H, CHNH), 3.90-3.95 (m, 2H, CH2OP), 4.09 (s, 5H, C5H5), 4.12 (s, 3H, C5H4), 4.18 (s, 1H, C5H4), 7.35–7.38 (m,
6H, m- and p-protons of phenyls +5H, C6H5), 7.92 (m, 4H, o-protons of phenyls); 13C
NMR (CDCl3, ppm): δ 8.21 (CH3 of Cp*(C5Me5)), 46.26 (CH2NH), 62.14 (d, 3J = 7.0
Hz, CHNH), 67.65, 67.90, 68.04, 68.40, 68.75, 70.64 (C5H4+C5H5+CH2OP), 86.06
(i-C5H4), 94.11 (d, 2J=3.0 Hz, C5Me5), 127.75, 127.85, 127.94, (C6H5), 128.47 (s,
carbons of phenyls), 130.95 (d, J = 6.7 Hz, m-carbons of phenyls), 133.10 (d, J = 11.5 Hz,
o-carbons of phenyls), 135.75 (d, J = 60.3 Hz, i-carbons of phenyls), 139.78 (i-C6H5);
31
P-{1H} NMR (CDCl3, ppm): δ 74.08 (s, O-P-(Ph)2); IR (KBr pellet in cm-1) υ: (C-Cp):
3054, (C=C-Cp): 1451, (P-Ph): 1436, (O–P): 1023; Anal. Calcd for [C41H45NOPFeIrCl2]
29
4.8.2. [(2S)-2-(Ferrocenylmethylamino)-2-phenylethyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (2a)
Fe N H O Ph PPh2 Fe N H O Ph PPh2 Ir Cl Cl Toluene 1/2[Ir(5-C5Me5)(-Cl)Cl]2 Yield: 0.152 g, 86.0 %; mp:131-1330C; [α]D20 = +15.8 o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.38 (d, 15H, 4J=2.0 Hz, CH3 of Cp* (C5Me5)), 3.25 (d, J=12.6 Hz, 1H, CH2NH, (a)), 3.46 (br, 1H, CH2NH, (b)), 3.87 (br, 1H, CHNH), 3.95 (br, 2H, CH2OP), 4.09 (s, 5H, C5H5), 4.13 (s, 3H, C5H4), 4.23 (s, 1H, C5H4), 7.31–7.39 (m, 6H, m- and p-protons of phenyls +5H, C6H5), 7.90 (d, J=7.5 Hz, 4H, o-protons of phenyls); 13C
NMR (CDCl3, ppm): δ 8.26 (CH3 of Cp* (C5Me5)), 46.17 (CH2NH), 62.06 (d, 3J = 8.0
Hz, CHNH), 67.82, 68.10, 68.12, 68.17, 68.45, 68.62 (C5H4+C5H5+CH2OP), 86.61
(i-C5H4), 94.14 (d, 2J=3.0 Hz, C5Me5), 127.72, 127.82, 127.93, (C6H5), 128.57 (s,
carbons of phenyls), 131.06 (d, J = 15.1 Hz, m-carbons of phenyls), 132.95 (br, o-carbons of phenyls), 135.70 (d, J=61.4 Hz, i-carbons of phenyls), 141.54 (i-C6H5); 31P-{1H}
NMR (CDCl3, ppm): δ 74.24 (s, O-P-(Ph)2); IR (KBr pellet in cm-1) υ: (C-Cp): 3058,
(C=C-Cp): 1451, (P-Ph): 1436, (O–P): 1023; Anal. Calcd for [C41H45NOPFeIrCl2]
4. MATERIALS and METHODS
30
4.8.3. [(2R)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (3a)
Fe N H O PPh2 Ir Cl Cl Ph Fe N H O PPh2 Ph Toluene 1/2[Ir(5-C5Me5)(-Cl)Cl]2 Yield: 0.149 g, 85.3 %; mp:125-126oC); [α]D20 = +9.5o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.38 (d, 15H, 4J=2.1Hz, CH3 of Cp* (C5Me5)), 2.82 (br, 1H, CH2C6H5 (a)), 2.86 (m, 1H, CH2C6H5 (b)), 3.03 (br, 1H, CHNH), 3.38 (br, 1H, CH2NH, (a)), 3.46 (m, 1H, CH2NH, (b)), 3.83 (br, 2H, CH2OP), 4.01 (s, 5H, C5H5), 4.07 (s, 3H, C5H4), 4.12
(s, 1H, C5H4), 7.12–7.40 (m, 6H, m- and p-protons of phenyls +5H, CH2C6H5) 7.95-8.02
(m, 4H, o-protons of phenyls); 13C NMR (CDCl3, ppm): δ 8.22 (CH3 of Cp* (C5Me5)),
37.97 (CH2Ph), 46.51 (CH2NH), 58.66 (d, 3J = 7.0 Hz, CHNH), 67.84, 68.40, 68.63,
68.80, 69.07, 69.66 (C5H4+C5H5 +CH2OP), 86.51 (i-C5H4), 94.10 (d, 2J=3.0 Hz, C5Me5),
126.51, 128.60, 129.31, (CH2C6H5), 127.81 (d, J = 11.1 Hz, m-carbons of phenyls),
132.61 (d, J = 11.1 Hz, p-carbons of phenyls), 133.69 (d, J = 11.1, o-carbons of phenyls), 136.17 (br, i-carbons of phenyls), 138.16 (i-CH2C6H5); 31P-{1H} NMR (CDCl3, ppm): δ
73.72 (s, O-P-(Ph)2); IR (KBr pellet in cm-1) υ: (C-Cp): 3058, (C=C-Cp): 1451, (P-Ph):
1436, (O–P): 1023; Anal. Calcd for [C42H47NOPFeIrCl2] (931.78 g/mol): C, 54.14; N,
31
4.8.4.[(2S)-2-(Ferrocenylmethylamino)-3-phenylpropyldiphenylphosphinito (dichloro(ɳ5-pentamethylcyclopentadienyl)iridium(III))] (4a)
Fe N H O PPh2 Ir Cl Cl Ph Fe N H O PPh2 Ph Toluene 1/2[Ir(5-C5Me5)(-Cl)Cl]2 Yield: 0.143 g, 81.9 %; mp:129-130oC; [α]D20 = -9.5o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.38 (d, 15H, 4J=1.3 Hz, CH3 of Cp* (C5Me5)), 2.77-2.82 (br, 2H CH2C6H5), 3.05 (br, 1H, CHNH), 3.48 (br, 2H, CH2NH), 3.84 (br, 2H, CH2OP), 4.02 (s, 5H, C5H5), 4.08 (s, 3H, C5H4), 4.14 (s, 1H, C5H4), 7.11–7.40 (m, 6H, m- and p-protons of
phenyls +5H, CH2C6H5) 7.93-8.01 (m, 4H, o-protons of phenyls); 13C NMR (CDCl3,
ppm): δ 8.23 (CH3 of Cp* (C5Me5)), 38.06 (CH2Ph), 46.53 (CH2NH), 58.65 (d, 3J = 6.0
Hz, CHNH), 67.82, (2J =7.0 Hz, CH2OP), 67.91, 68.40, 68.62 68.79, 69.66
(C5H5+C5H4), 86.65 (i-C5H4), 94.10 (d, 2J=2.0 Hz, C5Me5), 126.51, 128.60, 129.31,
(CH2C6H5), 127.80 (d, J = 11.1 Hz, m-carbons of phenyls), 132.63 (d, J = 11.1 Hz, p-carbons of phenyls), 133.68 (d, J = 12.1, o-carbons of phenyls), 136.17 (br, i-carbons of
phenyls), 138.17 (i-CH2C6H5); 31P-{1H} NMR (CDCl3, ppm): δ 73.85 (s, O-P-(Ph)2 IR
(KBr pellet in cm-1) υ: (C-Cp): 3054, (C=C-Cp): 1449, (P-Ph): 1436, (O–P): 1023; Anal. Calcd for [C42H47NOPFeIrCl2] (931.78 g/mol): C, 54.14; N, 1.50; H, 5.08; Found: C,
4. MATERIALS and METHODS
32
4.8.5.(1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyl diphenylphosphinito[dichloro(η6-p-cymene)ruthenium(II)] (5a)
N Ph Ph Ph2PO Ru Cl Cl N Ph Ph Ph2PO THF 1/2[Ru(-p-cymene)Cl2]2 At first, [Ru(η6
-p-cymene)(µ-Cl)Cl]2 (0.05 g, 0.09 mmol) and
(1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyldiphenylphosphinit e, (5) (0.10 g, 0.18 mmol) were dissolved in 30 mL of thf under an argon atmosphere and stirred for 30 minute at room temperature. The volume was concentrated to ca. 1–2 mL under reduced pressure and addition of petroleum ether (25 mL) gave 5a as a red solid. The product was collected by filtration and dried in vacuo (yield: 0.13 g, 85.8 %; mp: 123-124 0C); [α]D25 = + 27o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.05 (m, 6H,
CH3)2CHPh of p-cymene); 1.20 (d, J = 7.9 Hz, 3H, -CHCH3), 1.49 (s, 3H, CH3Ph of p-cymene), 2.37 (m, 1H, –CH- of p-cymene), 2.59 (m, 1H, -CH2N (a)), 2.87 (dd (pseudo
t), J = 11.6 Hz, J = 11.5 Hz 1H, -CH2N (b)), 3.20 (d, J = 13.3 Hz, 1H, -CH2Ph, (a)), 3.38
(d, J = 13.2 Hz, 1H, -CH2Ph, (b)), 4.25 (q, J = 6.5 Hz, 1H, -CHCH3), 4.47 (d, J = 5.8 Hz,
1H, aromatic proton of p-cymene), 4.84 (d, J = 6.0 Hz, 1H, aromatic proton of p-cymene), 4.96 (d, J = 6.0 Hz, 1H, aromatic proton of p-cymene), 5.17 (d, J = 5.8 Hz, 1H, aromatic proton of p-cymene), 5.41 (m, 1H, CHPh), 6.65-7.07 (m, 8H, Ar-H), 7.29-7.37 (m, 12 H, Ar-H), 7.57 (t, J = 8.3 Hz, 3H, Ar-H), 7.88 (t, J = 7.8 Hz, 4H, Ar-H); 13C NMR (CDCl3,
ppm): δ 10.51 (CHCH3), 17.01 (CH3Ph’ı of p-cymene), 21.99 ((CH3)2CHPh of p-cymene), 29.80 (–CH- of p-cymene), 52.22 (CHCH3), 54.54, 55.39 (CH2Ph, CH2N),
77.24 (CHPh), 87.16, 88.23, 88.56, 89.00 (aromatic carbons of p-cymene), 99.54, 111.76 (quaternary carbons of p-cymene), 124.63, 124.78, 126.91, 126.97, 127.09, 127.72, 127.82, 130.00, 130.73, 130.83, 132.13, 132.38, 132.49, 133.67, 138.47, 138.97, 140.13 (aromatic carbons), 124.18 (d, J = 4.0 Hz, m-carbons of OPPh2), 125.19 (s, p-carbons of
33
PPh2), 127.36 (d, J = 6.0 Hz, o-carbons of OPPh2), 134.25 (d, J = 11.1 Hz, i-carbons of
OPPh2); 31P-{1H} NMR (CDCl3, ppm): δ 109.70 (s, O-PPh2); IR (cm-1): υ 3057
(aromatic C-H), 2963 (aliphatic C-H), 1435 (P-Ph), 970 (O-P), 531 (Ru-P); Anal. Calc. for C49H50NOPRuCl2 (871.89 g/mol): C 67.50, H 5.78, N 1.61; found C 67.41, H 5.70, N
1.52.
4.8.6.(1R)-2-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-1-phenylethyl diphenylphosphinito[dichloro(η6-benzene)ruthenium (II)] (5b)
N Ph Ph Ph2PO Ru Cl Cl N Ph Ph Ph2PO THF 1/2[Ru(-benzene)Cl2]2
The protocol for 5a was employed. Yield: 0.12 g, 83.9 %; mp: 191-193 0C); [α]D25
= + 113o (c 1, CH2Cl2). 1H NMR (CDCl3, ppm): δ 1.17 (d, J = 6.6 Hz, 3H, -CHCH3);
2.32 (d, J = 10.0 Hz, 1H, -CH2N (a)), 2.88 (dd (pseudo t), J = 11.5 Hz, J = 11.3 Hz, 1H,
-CH2N (b)), 3.14 (d, J = 13.2 Hz, 1H, -CH2Ph, (a)), 3.39 (d, J = 13.2 Hz, 1H, -CH2Ph,
(b)), 4.27 (q, J = 6.6 Hz, 1H, -CHCH3), 4.95 (s, 6H, aromatic protons of benzene),
5.63-5.70 (m, 1H, CHPh), 6.73-7.58 (m, 23H, Ar-H), 7.96 (t, J = 8.5 Hz, 2H, Ar-H), 8.06 (t, J = 8.9 Hz, 2H, Ar-H); 13C NMR (CDCl3, ppm): δ 10.61 (CHCH3), 51.99 (CHCH3),
54.98, 55.44 (CH2Ph, CH2N), 77.23 (CHPh), 90.13 (d, J = 4.0 Hz, aromatic carbons of
benzene), 124.71, 124.82, 126.85, 126.99, 127.16, 127.59, 127.79, 127.92, 128.12, 130.06, 130.42, 130.70, 131.48, 133.68, 138.37, 138.72, 140.44 (aromatic carbons), 124.07 (d, J = 7.0 Hz, m-carbons of OPPh2), 125.13 (s, p-carbons of PPh2), 127.46 (d, J =
6.0 Hz, o-carbons of OPPh2), 135.42 (d, J = 13.1 Hz, i-carbons of OPPh2); 31P-{1H}
NMR (CDCl3, ppm): δ 104.76 (s, O-PPh2); IR (cm-1): υ 3049 (aromatic C-H), 2966,
2935, 2888, 2841, 2806 (aliphatic C-H), 1436 (P-Ph), 992 (O-P), 524 (Ru-P); Anal. Calc. for C45H42NOPRuCl2 (815.78 g/mol): C 66.26, H 5.19, N 1.72; found C 66.18, H 5.10, N
4. MATERIALS and METHODS
34
4.8.7.(2S)-1-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-3-phenoxy propan-2-yldiphenylphosphinito[dichloro(η6-p-cymene)
ruthenium (II)] (6a)
N Ph Ph2PO Ru Cl Cl N Ph Ph2PO THF 1/2[Ru(-p-cymene)Cl2]2 OPh OPh
The protocol for 5a was employed. Yield: 0.13 g, 85.9 %; mp: 168-170 0C); [α]D25
= + 56o (c 1, CH2Cl2). 1H NMR (CDCI3, ppm): δ 0.93 (d, J = 6.9 Hz, 3H, CH3)2CHPh of p-cymene), 1.09 (d, J = 6.9 Hz, 3H, CH3)2CHPh of p-cymene), 1.26 (d, J = 6.7 Hz, 3H,
-CHCH3), 1.70 (s, 3H, CH3Ph of p-cymene), 2.30 (m, 2H, CH2N), 2.50 (m, 1H, –CH- of p-cymene), 2.61 (t, J = 9.3 Hz, 1H, CH2OPh (a)), 3.41 (d, J = 13.4 Hz, 1H, CH2Ph, (a)),
3.60 (d, J = 10.4 Hz, 1H, CH2OPh (b)), 3.71 (d, J = 13.4 Hz, 1H, CH2Ph, (b)), 4.47 (q, J =
6.6 Hz, 1H, CHCH3), 4.75 (br, 1H, CHCH2), 5.22 (d, J = 5.8 Hz, 1H, aromatic proton of p-cymene), 5.35 (d, J = 5.8 Hz, 1H, aromatic proton of p-cymene), 5.48 (q, J = 5.8 Hz,
2H, aromatic protons of p-cymene), 5.98 (d, J = 8.0 Hz, 2H, Ar-H), 6.91 (t, J = 7.3 Hz, 1H, Ar-H), 7.11-7.82 (m, 22H, Ar-H), 7.99 (t, J = 8.7 Hz, 2H, Ar-H); 13C NMR (CDCl3,
ppm): δ 8.79 (-CHCH3), 17.00 (CH3Ph of p-cymene), 21.60, 22.14 ((CH3)2CHPh of p-cymene), 29.98 (–CH- of p-cymene), 49.20 (CH2N), 51.90 (CHCH3), 56.14 (CH2Ph),
69.29 (-CH2OPh), 75.91 (d, J = 6.0 Hz, -CHCH2), 86.19 (d, J = 6.0 Hz, aromatic carbon
of p-cymene), 87.94 (d, J = 5.0 Hz, aromatic carbon of p-cymene), 89.64 (s, aromatic carbon of p-cymene), 92.29 (d, J = 5.0 Hz, aromatic carbon of p-cymene), 97.51, 111.20 (quaternary carbons of p-cymene), 113.75, 120.38, 124.73, 125.01, 125.20, 125.41, 127.22, 127.33, 127.90, 129.20, 130.27, 131.96, 132.12, 133.88, 134.17, 138.51, 139.13, 158.26 (aromatic carbons), 127.82 (d, J = 10.1 Hz, m-carbons of OPPh2), 128.07 (s, p-carbons of OPPh2), 130.65 (d, J = 15.1 Hz, o-carbons of OPPh2), (not observed i-carbons of OPPh2); 31P-{1H} NMR (CDCl3, ppm): δ 111.15 (s, O-PPh2); IR (cm-1): υ
35
(Ru-P); Anal. Calc. for C50H52NO2PRuCl2 (901.92 g/mol): C 66.59, H 5.81, N 1.55;
found C 66.42, H 5.75, N 1.49.
4.8.8.(2S)-1-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino}-3-phenoxypropan -2-yl diphenylphosphinito[dichloro(η6-benzene)ruthenium (II)] (6b)
N Ph Ph2PO Ru Cl Cl N Ph Ph2PO THF 1/2[Ru(-benzene)Cl2]2 OPh OPh
The protocol for 5a was employed. Yield: 0.12 g, 85.6 %; mp: 147-149 0C); [α]D25
= + 90o (c 1, CH2Cl2). 1H NMR (CDCI3, ppm): δ 1.27 (d, J = 6.6 Hz, 3H, -CHCH3);
2.26 (d, J = 9.5 Hz, 1H, CH2N (a)), 2.39 (dd (pseudo t), J = 11.8 Hz, J = 11.7 Hz 1H,
CH2N (b)), 2.68 (t, J = 9.1 Hz, 1H, CH2OPh (a)), 3.49 (d, J = 13.4 Hz, 1H, CH2Ph, (a)),
3.65 (d, J = 9.6 Hz, 1H, CH2OPh (b)), 3.92 (d, J = 13.4 Hz, 1H, CH2Ph, (b)), 4.56 (q, J =
6.3 Hz, 1H, CHCH3), 4.94 (m, 1H, CHCH2), 5.47 (s, 6H, aromatic protons of benzene),
5.98 (d, J = 8.0 Hz, 2H, Ar-H), 6.90 (t, J = 7.2 Hz, 1H, Ar-H ),7.09-7.94 (m, 24H, Ar-H);
13
C NMR (CDCl3, ppm): δ 9.09 (-CHCH3), 49.19 (CH2N), 52.47 (CHCH3), 56.40
(CH2Ph), 69.04 (-CH2OPh), 76.74 (d, J = 3.0 Hz, -CHCH2), 90.32 (d, J = 4.0 Hz,
aromatic carbons of benzene), 113.82, 120.52, 124.71, 125.00, 125.24, 125.44, 127.22, 127.98, 129.21, 130.30, 130.64, 131.18, 132.15, 133.92, 134.62, 138.45, 139.25, 158.18 (aromatic carbons), 127.46 (d, J = 10.1 Hz, m-carbons of OPPh2), 128.10 (s, p-carbons of
OPPh2), 128.28 (d, J = 22.1 Hz, o-carbons of OPPh2), 140.19 (d, J = 58.4 Hz, i-carbons of
OPPh2); 31P-{1H} NMR (CDCl3, ppm): δ 109.59 (s, O-PPh2); IR (cm-1): υ 3058
(aromatic C-H), 2966, 2935 (aliphatic C-H), 1435 (P-Ph), 967 (O-P), 537 (Ru-P); Anal. Calc. for C46H44NO2PRuCl2 (845.81 g/mol): C 65.32, H 5.24, N 1.66; found C 65.18, H
4. MATERIALS and METHODS
36 4.9.Catalytic Studies
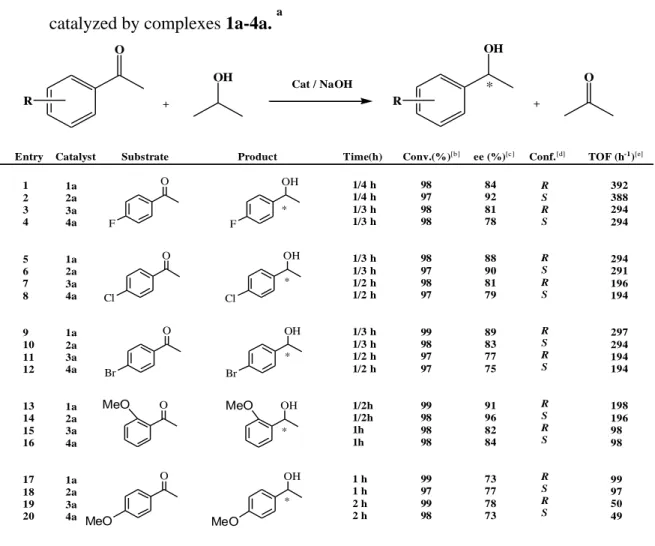
Within the scope of this thesis, catalytic activities of the ruthenium and iridium complexes 1a-4a in“asymmetric transfer hydrogenation reactions of ketones”were investigated and the results are shown in the Tables.
“Table 1. Asymmetric transfer hydrogenation of acetophenone with iso-PrOH catalysed by complexes 1a-4a.” O + OH OH + O cat. *
Entry Catalyst S/C/NaOH Time(h) Conversion(%)[k] % ee[l] Conf.[m] TOF(h-1)[n]
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 1a 2a 3a 4a 1a 2a 3a 4a 1a 2a 3a 4a 1a 1a 1a 1a 1a 2a 3a 4a 1a 2a 3a 4a 100:1:5 100:1:5 100:1:5 100:1:5 100:1 100:1 100:1 100:1 100:1:5 100:1:5 100:1:5 100:1:5 100:1:3 100:1:5 100:1:7 100:1:9 500:1:5 500:1:5 500:1:5 500:1:5 1000:1:5 1000:1:5 1000:1:5 1000:1:5 13 (49) 15 (52) 11 (33) 12 (35) <5 <5 <5 <5 99 (97) 95 (93) 99 (97) 98 (96) 94 99 90 88 98 99 99 99 98 98 99 99 85 (81) 90 (88) 79 (78) 76 (75) .... .... .... .... 94 (90) 95 (91) 81 (78) 77 (74) 92 94 90 91 91 96 80 78 83 91 81 76 24 (72) 24 (72) 24 (72) 24 (72) 1 1 1 1 1/3 (1/3) 1/3 (1/3) 1/2 (1/2) 1/2 (1/2) 1/3 1/3 1/3 1/3 3/2 3/2 2 2 3 3 5 5 [a] [a] [a] [a] R S R S .... .... .... .... R S R S R R R R R S R S R S R S <5 <5 <5 <5 .... .... .... .... 297(291) 285(279) 198(194) 196(192) 282 297 270 264 327 330 248 248 327 327 198 198 [e] [f] [g] [h] [c] [c] [c] [c] [b] [b] [b] [b] [d] [d] [d] [d] [i] [i] [i] [i] [j] [j] [j] [j] “
“ Reaction conditions:a At room temperature; acetophenone/Cat./NaOH, 100:1:5; b Refluxing in iso-PrOH; acetophenone/Cat.100:1, in the absence of base; c Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 100:1:5; d Refluxing in iso-PrOH; acetophenone/ Cat./KOH, 100:1:5; e Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 100:1:3; f Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 100:1:5; g Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 100:1:7; h Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 100:1:9; i Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 500:1:5; j Refluxing in iso-PrOH; acetophenone/ Cat./NaOH, 1000:1:5; kDetermined by GC (three independent catalytic experiments); l Determined by capillary GC analysis using a chiral cyclodex B (Agilent) capillary column;m Determined by comparison of the retention times of the enantiomers on the GC traces with literature values, an (S)- or (R)-configuration was obtained in all experiments; n TOF = (mol product/mol cat.)x h-1.””
![Figure 10. A is accurate MS1 spectrum of N-benzyl-N-[(S)-1-[α-naphthylethyl]amine, B and C](https://thumb-eu.123doks.com/thumbv2/9libnet/3277115.9175/70.893.123.687.142.702/figure-accurate-ms-spectrum-n-benzyl-naphthylethyl-amine.webp)
![Figure 12. A is accurate MS1 spectrum of (2S)-1-{benzyl[(1S)-1-(naphthalen-1-yl)ethyl]amino 3-phenoxypropan-2-ol, B and C are the spectra that show the measured and predicted exact mass of the compound (2S)-1-{benzyl[(1S)-1-(naphthalen-1-yl)e](https://thumb-eu.123doks.com/thumbv2/9libnet/3277115.9175/72.893.124.686.126.912/accurate-spectrum-naphthalen-phenoxypropan-measured-predicted-compound-naphthalen.webp)
